Case
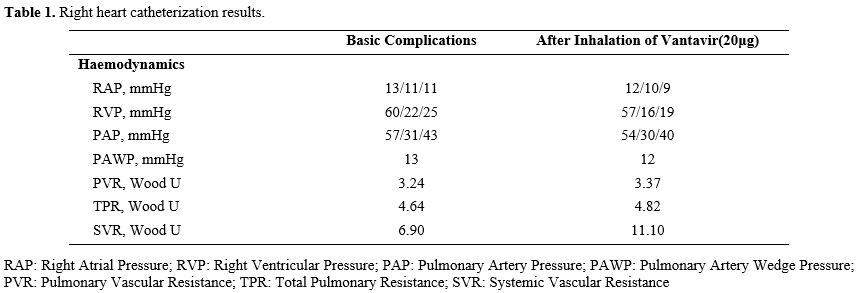
We present the case of a 33-year-old Chinese man diagnosed with β-thalassemia at the age of 1. He has been receiving intermittent transfusion therapy and regular iron-chelating therapy since the age of 30. His genotype is CD17(A〉T)/βE, and he underwent splenectomy at age 11. Due to limited blood resource availability in the local healthcare system during the early treatment phase, which constrained consistent transfusion access, he received occasional transfusions every 6-10 months to maintain his hemoglobin levels at 70-80 g/L. From age 30, he also received daily iron-chelating therapy using Deferasirox (30 mg/kg/d). He was considered a NTDT patient.In February 2022, at age 31, he experienced chest discomfort and shortness of breath following physical activity, which resolved with rest. He was admitted to a cardiovascular hospital where initial tests revealed the following: white blood cell count (WBC) 11.29×10^9/L, hemoglobin (Hb) 75 g/L, platelet count (PLT) 671×10^9/L, nucleated red blood cell count (NRBC) 11.03×10^9/L, serum ferritin (SF) 1238.7 ng/mL, prothrombin time (PT) 14.8 s, and D-dimer 0.33 µg/mL. Liver function tests showed total bilirubin (TBil) 102.12 µmol/L, indirect bilirubin (IBil) 80.95 µmol/L, and lactic dehydrogenase (LDH) 174 U/L. Echocardiography demonstrated mild tricuspid regurgitation, with a tricuspid regurgitation jet velocity (TRV) of 3.4 m/s and an estimated pulmonary artery systolic pressure of 51 mmHg. Pulmonary function tests showed moderate to severe restrictive ventilatory dysfunction; small airway dysfunction; pulmonary diffusing capacity was moderately decreased, and bronchodilation tests were negative. Contrast-enhanced CT of the pulmonary arteries results showed no evidence of embolism in the segmental and proximal pulmonary arteries, and pulmonary artery dilatation is present. Right heart catheterization (Table 1) results showed pulmonary artery pressure (PAP) 57/31/43mmHg, pulmonary vascular resistance (PVR) 3.24 Wood U, and pulmonary artery wedge pressure (PAWP) 13mmHg, which indicated PAP and PVR, with a decreased PAWP, suggesting precapillary pulmonary hypertension. His 6-minute walk test (6MWT) recorded 356 meters.
 |
|
The patient was diagnosed with PAH and was treated with spironolactone for diuresis and digoxin to enhance cardiac function. Additionally, he received low-dose Riociguat (0.5 mg three times daily) to lower pulmonary artery pressure due to the following treatment-emergent headache. After approximately three months on this regimen, his condition showed no significant improvement. In June 2022, the 6MWT was 350 meters. Echocardiography demonstrated mild tricuspid regurgitation, with a TRV of 3.3 m/s and an estimated pulmonary artery systolic pressure of 49 mmHg.
Luspatercept was approved by China’s National Medical Products Administration (NMPA) in 2022 for the treatment of adult patients with β-thalassemia who require regular red blood cell transfusions and do not have contraindication in PAH. Then, the patient began receiving Luspatercept at 1mg/kg every 21 days, and after two months, the dose was adjusted to 1.25mg/kg every 21 days. In July 2022, the magnetic resonance imaging (MRI) results indicated Cardiac T2* MRI: 46.89 ms (normal >20 ms); Liver iron concentration: >14 mg/g dw (Severe overload). Figures 1 show the changes in serum ferritin and hemoglobin levels following the Luspatercept therapy. Figures 2 show the changes in the Echocardiogram before and after treatment with Luspatercept. The patient reported improved chest discomfort and shortness of breath after the second dose of Luspatercept. 2 months after receiving Luspatercept, he followed up with the hematological index (Table 2), indicating increased HB, decreased SF, and improved hemolysis markers (bilirubin and NRBC). At 14 and 21 months of follow-up, improvements were maintained, and echocardiography showed normal TRV (Table 2). After improving significantly, the patient enhanced their motivation to seek further treatment. This improvement in compliance, combined with the family’s efforts to identify alternative blood supply sources, eventually enabled more consistent transfusions. In April 2024, after detailed counseling, the patient made an informed decision to discontinue luspatercept treatment in order to enroll in a potentially curative gene therapy clinical trial, requiring ≥8-week washout of erythroid-active agents. An echocardiogram at the cardiovascular hospital in August 2024 revealed mild tricuspid regurgitation, with a TRV of 3 m/s and an estimated pulmonary artery systolic pressure of 41 mmHg.
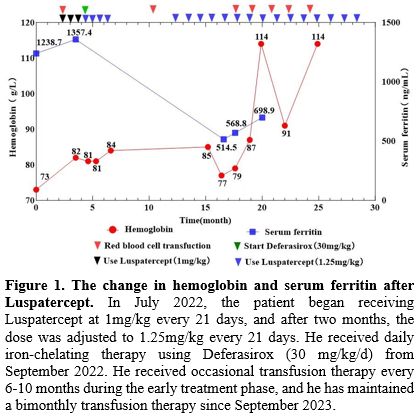 |
Figure 1. The change in hemoglobin and serum ferritin after Luspatercept. In July 2022, the patient began receiving Luspatercept at 1mg/kg every 21 days, and after two months, the dose was adjusted to 1.25mg/kg every 21 days. He received daily iron-chelating therapy using Deferasirox (30 mg/kg/d) from September 2022. He received occasional transfusion therapy every 6-10 months during the early treatment phase, and he has maintained a bimonthly transfusion therapy since September 2023. |
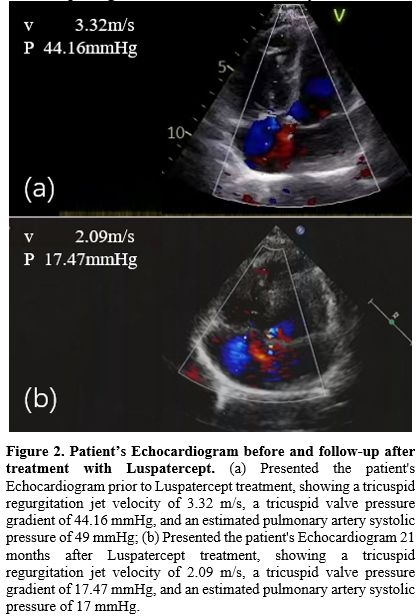 |
Figure 2. Patient’s Echocardiogram before and follow-up after treatment with Luspatercept.
(a) Presented the patient's Echocardiogram prior to Luspatercept
treatment, showing a tricuspid regurgitation jet velocity of 3.32 m/s,
a tricuspid valve pressure gradient of 44.16 mmHg, and an estimated
pulmonary artery systolic pressure of 49 mmHg; (b) Presented the
patient's Echocardiogram 21 months after Luspatercept treatment,
showing a tricuspid regurgitation jet velocity of 2.09 m/s, a tricuspid
valve pressure gradient of 17.47 mmHg, and an estimated pulmonary
artery systolic pressure of 17 mmHg. |
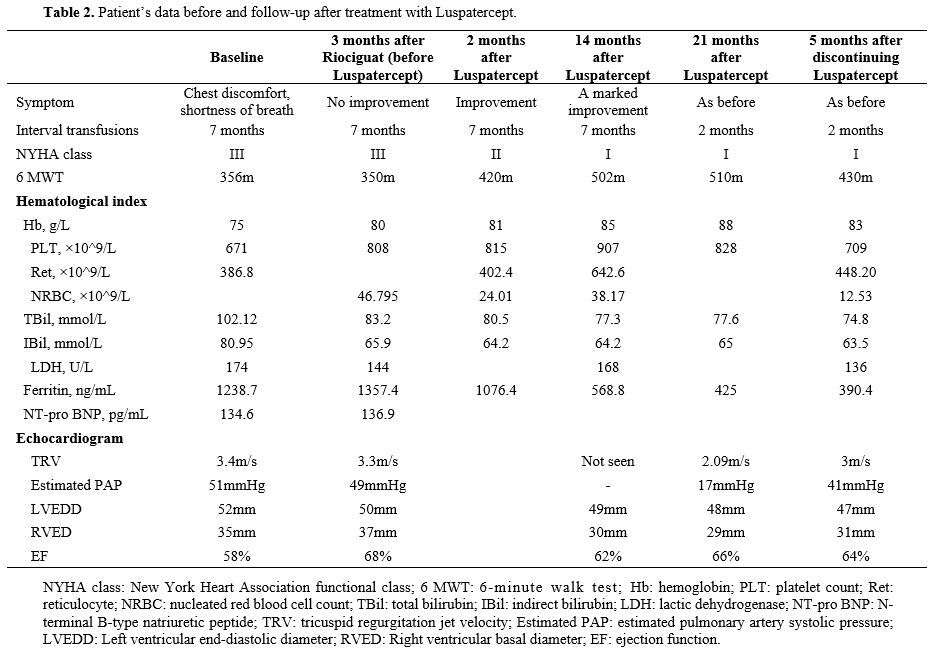 |
Table 2. Patient’s data before and follow-up after treatment with Luspatercept. |
Discussion
Based on the patient's Echocardiogram and Contrast-enhanced CT of the pulmonary arteries, pulmonary hypertension secondary to left heart disease, pulmonary hypertension associated with hypoxia, or pulmonary artery obstruction due to lung disease has been excluded. PAH secondary to β-thalassemia is classified under Group 5 pulmonary hypertension (PH), encompassing disorders with multifactorial and incompletely understood mechanisms.[6] The presented case highlights a splenectomized patient with β-thalassemia exhibiting hallmark features of chronic hemolysis, iron overload, platelet activation, and hypercoagulability - all recognized risk factors for PAH progression.Conventional management of β-thalassemia-associated PAH centers on transfusion regimens, iron chelation therapy, and supportive measures such as oxygen supplementation, anticoagulation, and cardiac function optimization.[7] According to current ESC/ERS PH Guidelines,[8] Sildenafil, bosentan, and other similar drugs are primarily indicated for pulmonary arterial hypertension (Group 1). However, no prospective studies have confirmed their efficacy in treating pulmonary arterial hypertension caused by thalassemia (Group 5). Despite these interventions, pulmonary vasodilators - including phosphodiesterase type 5 inhibitors (PDE-5i), endothelin receptor antagonists, and the soluble guanylate cyclase stimulator riociguat - demonstrated limited efficacy and unfavorable adverse effect profiles in this patient.[7] Notably, three months of low-dose riociguat failed to ameliorate symptoms or objective measures of PAH.
The 1.25 mg/kg dose of Luspatercept elicited a hemoglobin elevation of approaching 10 g/L, confirming its therapeutic efficacy for anemia management.[9] Intriguingly, concurrent enhancements in 6-minute walk test (6MWT) performance and tricuspid regurgitant velocity (TRV) on echocardiography suggested a rapid reduction in pulmonary vascular resistance. Discontinuation of luspatercept precipitated increased transfusion requirements and recrudescence of pulmonary hypertension, underscoring its therapeutic dependency. Hematologic profiling revealed diminished nucleated red blood cell (NRBC) counts post-treatment, indicative of attenuated ineffective erythropoiesis - a finding potentially linked to PAH mitigation, though mechanistic clarity remains elusive. We acknowledge that this confounding factor (concurrent transfusions) may complicate the interpretation of luspatercept’s isolated effects on PAH. However, the temporal association between luspatercept initiation and symptomatic improvement (prior to transfusion intensification) suggests a potential role of the drug, warranting further investigation in controlled settings.
In chronic hemolytic anemia, PAH pathogenesis is driven by nitric oxide (NO) depletion due to free hemoglobin-mediated scavenging, compounded by L-arginine metabolic dysregulation, endothelial dysfunction, and elevated endothelin-1 levels.[10] The phase 3 COMMANDS trial[11] further supports luspatercept’s cardioprotective role, demonstrating reduced NT-proBNP levels, likely mediated via TGF-β signaling suppression, apoptotic pathway modulation, and downregulation of pro-inflammatory mediators.[12,13] These effects may synergistically enhance NO bioavailability, offering a plausible pathway for PAH alleviation. Meantime, a phase II, open-label study of sotatercept revealed that sotatercept reduced transfusion requirements in TDT patients.[14] By sequestering SMAD2/3 pathway ligands (e.g., activins, growth differentiation factors), sotatercept restores balance between pro-proliferative and anti-proliferative signaling in the pulmonary vasculature.[15] Its efficacy in PAH is evidenced by a phase 3 trial showing superior 6MWT outcomes versus placebo (p<0.001).[16] As a similar agent to luspatercept,[17] which has demonstrated effectiveness in treating PAH in thalassemia in this case, the potential efficacy of sotatercept in PAH in thalassemia (classified under Group 5) is worth exploring.
β-thalassemia-related PAH confers significant mortality risk, yet pharmacologic interventions - particularly those achieving >25% hemodynamic improvement - may attenuate this burden.[4] In this case, luspatercept yielded dual hematologic and cardiopulmonary benefits, suggesting a novel role in modulating pulmonary vascular resistance. With symptomatic relief improving compliance, the patient could maintain transfusion regimens more effectively, thereby meeting the eligibility criteria for experimental gene therapy as a potential cure. While preliminary, these observations warrant rigorous investigation to delineate luspatercept’s pleiotropic effects and optimize therapeutic strategies in this high-risk population. At the same time, based on the role of microthrombotic events in the evolution of PAH in thalassemia, an increase in the thrombotic risk may become evident in the long-term use of luspatercept and must be promptly recognized.
Author Contributions
Xiaolin Yin, Yinjiang Tang, and Beibei Yang contributed to the design and data acquisition. Beibei Yang and Dongmei Liu contributed to the data analysis, discussion, and manuscript writing. Dongmei Liu and Changyu Yang contributed to imaging data acquisition and data analysis. Yali Zhou and Guiping Liao contributed to data acquisition. Jian Huang and Yingying Li contributed to scheduling patient follow-ups. All authors reviewed the manuscript.Data Availability Statement
The data are available from the corresponding author upon reasonable request.References
- Taher AT, Musallam KM, Karimi M, El-Beshlawy A,
Belhoul K, Daar S, et al. Overview on practices in thalassemia
intermedia management aiming for lowering complication rates across a
region of endemicity: The optimal care study. Blood 2010;115:1886-92. https://doi.org/10.1182/blood-2009-09-243154 PMid:20032507
- Benza
RL, Miller DP, Barst RJ, Badesch DB, Frost AE, Mcgoon MD. An Evaluation
of Long-term Survival From Time of Diagnosis in Pulmonary Arterial
Hypertension From the REVEAL Registry n.d. https://doi.org/10.1378/chest.11-1460.
- Derchi
G, Galanello R, Bina P, Cappellini MD, Piga A, Lai ME, et al.
Prevalence and risk factors for pulmonary arterial hypertension in a
large group of β-thalassemia patients using right heart
catheterization: A webthal study. Circulation 2014;129:338-45. https://doi.org/10.1161/CIRCULATIONAHA.113.002124 PMid:24081970
- Ferrari B, Peyvandi F. How I treat thrombotic thrombocytopenic purpura in pregnancy. Blood 2020;136:2125-32. https://doi.org/10.1182/blood.2019000962 PMid:32797178
- Angelucci E. A new medical therapy for anemia in thalassemia. Blood 2019;133:1267-8. https://doi.org/10.1182/blood-2019-01-897587 PMid:30898772
- Humbert
M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022
ESC/ERS Guidelines for the diagnosis and treatment of pulmonary
hypertension. Eur Heart J 2022;43:3618-731. https://doi.org/10.1093/eurheartj/ehac237 PMid:36017548
- Fraidenburg DR, Machado RF. Pulmonary hypertension associated with thalassemia syndromes. Ann N Y Acad Sci 2016;1368:127-39. https://doi.org/10.1111/nyas.13037 PMid:27008311 PMCid:PMC4870173
- Humbert
M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022
ESC/ERS Guidelines for the diagnosis and treatment of pulmonary
hypertension. Eur Heart J 2022;43:3618-731. https://doi.org/10.1093/eurheartj/ehac237 PMid:36017548
- Taher
AT, Cappellini MD, Kattamis A, Voskaridou E, Perrotta S, Piga AG, et
al. Luspatercept for the treatment of anaemia in
non-transfusion-dependent β-thalassaemia (BEYOND): a phase 2,
randomised, double-blind, multicentre, placebo-controlled trial. Lancet
Haematol 2022;9:e733-44. https://doi.org/10.1016/S2352-3026(22)00208-3 PMid:36007538
- Morris
CR, Kim HY, Klings ES, Wood J, Porter JB, Trachtenberg F, et al.
Dysregulated arginine metabolism and cardiopulmonary dysfunction in
patients with thalassaemia. Br J Haematol 2015;169:887-98. https://doi.org/10.1111/bjh.13452 PMid:25907665 PMCid:PMC4452408
- Platzbecker
U, Della Porta MG, Santini V, Zeidan AM, Komrokji RS, Shortt J, et al.
Efficacy and safety of luspatercept versus epoetin alfa in
erythropoiesis-stimulating agent-naive, transfusion-dependent,
lower-risk myelodysplastic syndromes (COMMANDS): interim analysis of a
phase 3, open-label, randomised controlled trial. The Lancet
2023;402:373-85. https://doi.org/10.1016/S0140-6736(23)00874-7 PMid:37311468
- Sheida
Hayati, Amer M. Zeidan, Guillermo Garcia-Manero, Uwe Platzbecker, Aarif
Ahsan, Amit K. Verma, Srinivas Aluri, Manuel Ugidos Guerrero, Anita K.
Gandhi, Rajasekhar N.V.S. Suragani, Sadanand Vodala. Luspatercept
modulates inflammation in the bone marrow, restores effective
erythropoiesis/ hematopoiesis, and provides sustained clinical benefit
versus epoetin alfa: biomarker analysis from the phase 3 COMMANDS
study. Presented at the American Society of Hematology (ASH) Annual
Meeting 2023. https://doi.org/10.1182/blood-2023-178674
- Maroof
Hasan. Luspatercept reduces clonal hematopoiesis-associated cardiac
stress via modulation of mTORC1 and inflammatory signaling pathways.
Presented at the American Society of Hematology (ASH) Annual Meeting
2024 2024. https://doi.org/10.1182/blood-2024-194241
- Cappellini
MD, Porter J, Origa R, Forni GL, Voskaridou E, Galactéros F, et al.
Sotatercept, a novel transforming growth factor β ligand trap, improves
anemia in β-thalassemia: A phase II, open-label, dose-finding study.
Haematologica 2019;104:477-84. https://doi.org/10.3324/haematol.2018.198887 PMid:30337358 PMCid:PMC6395345
- Humbert
M, McLaughlin V, Gibbs JSR, Gomberg-Maitland M, Hoeper MM, Preston IR,
et al. Sotatercept for the Treatment of Pulmonary Arterial
Hypertension. New England Journal of Medicine 2021;384:1204-15. https://doi.org/10.1056/NEJMoa2024277 PMid:33789009
- Hoeper
MM, Badesch DB, Ghofrani HA, Gibbs JSR, Gomberg-Maitland M, McLaughlin
V V., et al. Phase 3 Trial of Sotatercept for Treatment of Pulmonary
Arterial Hypertension. New England Journal of Medicine
2023;388:1478-90. https://doi.org/10.1056/NEJMoa2213558 PMid:36877098
- Brancaleoni V, Nava I, Delbini P, Duca L, Motta I. Activin receptor-ligand trap for the treatment of β-thalassemia: A serendipitous discovery. Mediterr J Hematol Infect Dis 2020;12. https://doi.org/10.4084/mjhid.2020.075 PMid:33194149 PMCid:PMC7643807