In mammals iron is a constituent of many proteins, either as iron sulfur clusters (such as in the respiratory complexes I-III, mitochondrial aconitase, coenzyme Q10, DNA primase) or as heme (such as in hemoglobin, myoglobin, cytochrome proteins, myeloperoxidase, nitric oxide synthetases) or as constituent of other functional groups (such as hypoxia inducible prolyl hydroxylases). These iron-containing proteins are involved in cellular and organismal functions of vital importance, such as oxygen transport, mitochondrial respiration, DNA replication and repair, immunity, and cell signaling. During evolution, the great oxygenation event occurring 2.2-2.3 billion years ago caused oxygenation of the Earth’s atmosphere; this event elicited the oxidation of soluble Fe2+ to insoluble Fe3+. This event rendered iron less available for biological systems. It required the development of specialized systems for iron uptake, transport, recycling, storage, and export, and for protection from the toxic compounds that can be generated during redox cycling of iron in the presence of oxygen.[2] To prevent diseases related to iron deficiency or overload, iron homeostasis must be carefully regulated. Intracellular iron levels are controlled by the Iron Regulatory Element-Iron Regulatory Protein (IRE-IRP) system, while systemic iron availability is primarily adjusted to the body's needs through the Hepcidin-Ferroportin (FPN) axis.[3]
Dietary Iron Absorption
Mammals require small amounts of iron, which is essential for many biological activities and particularly for oxygen transport.The human body contains ~3-5 g of iron; most is present as heme in hemoglobin of erythroid cells (>2g) or myoglobin of muscles (~300 mg). The iron contained in hemoglobin is recycled in the process of erythrophagocytosis by reticuloendothelial macrophages. Macrophages in the spleen, liver, and bone marrow maintain a transient fraction of iron (~600 mg), while excess of the metal is stored in the liver parenchyma within ferritin (~1000 mg). The circulating pool of iron is small (2-4 mg) and is renewed every few hours to meet a daily iron requirement of about 20-25 mg. All other iron-containing proteins and enzymes contain only a minority of total iron (~8mg of iron) (Figure 1).[4]
 |
|
Systemic iron homeostasis requires the coordinated activity of different compartments involved in iron absorption, transport, storage, recycling, and high-level utilization.
The absorption of dietary iron is mediated by the duodenum and involves the absorption of free iron and iron contained in heme. The average daily requirement of approximately 20-25 mg of iron derives from dietary intake from heme iron present in meat and nonheme iron present in vegetables (1-2 mg) and mainly from the degradation of senescent RBCs (20-25 mg). The level of iron absorbed by the small intestine is low under physiological conditions but increases in conditions of iron deprivation.[4]
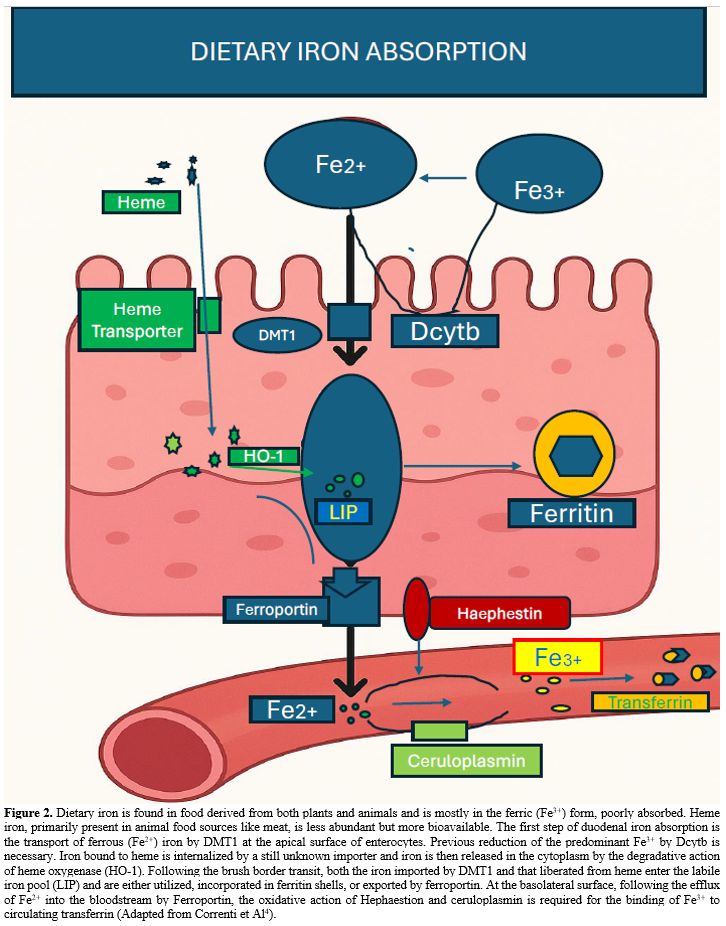
Most of the iron contained in food is Fe3+ and must be reduced to Fe2+ before its absorption, a function mediated by the ferric reductases Dcytb (duodenal cytochrome B). Dcytb is an integral membrane protein localized in the apical side of enterocytes that catalyzes the reduction of nonheme Fe3+ by electron transfer from ascorbate across the membrane. The activity of Dcytb is essential for iron absorption through divalent metal transporter-1 (DMT1) (Figure 2).
 |
|
Intestinal iron absorption involves three phases: apical uptake, enterocytic intracellular phase, and basolateral transfer.[4,5] The apical uptake consists of iron transport across the brush border and is mediated through the transport of Fe2+ across the apical membrane of enterocytes by DMT1. DMT1 activity in this process is essential, and mice lacking intestinal DMT1 develop a marked microcytic anemia and have reduced iron stores.[6] DMT1 mutations represent a rare cause of hypochromic anemias in humans.
In addition to facilitating ferrous iron absorption through the apical membrane of enterocytes, DMT1 also plays a role in recovering iron from urine via renal tubules and in releasing iron into the cytosol from acidified endosomes after diferric-transferrin-transferrin receptor internalization. DMT1 is regulated by iron itself: the 3’ untranslated regions of DMT1 mRNA may contain or lack an iron-responsive element (IRE), resulting in two different isoforms; the DMT1-IRE isoform is mainly expressed in enterocytes, and its expression is influenced by iron levels, with iron deficiency particularly causing an increase in DMT1 mRNA levels to promote stabilization.
Heme contained in foods rich in myoglobin or hemoglobin is absorbed by the duodenum through a still elusive membrane heme transporter. The mechanism of intestinal heme-bound iron absorption was not fully elucidated. Three heme transporters have been identified and seem to play a relevant role in maintaining heme homeostasis: protein-coupled folate transporter/heme carrier protein 1 (HCP1), heme responsive gene 1 (HRG1) and FLVCR1 (Feline Leukemia Virus type C Receptor 1) highly expressed in tissues that transport heme (intestine or hepatic cells) or synthesize high levels of heme such as erythroid cells (Figure 2).[7]
The enterocyte intracellular phase involves the transport of Fe2+ internalized into enterocytes, involving first the passage into the cytosolic labile iron pool (LIP), followed by either the utilization of this iron or its incorporation into ferritin or its exportation out of enterocytes. The level of ferritin present in enterocytes is regulated according to intracellular iron content through the IRE-IRP system, being decreased by iron deprivation and increased by iron excess.
The last step of intestinal iron absorption involves the efflux of Fe2+ at the basolateral membrane, which is mediated by the membrane transporter ferroportin (FPN1). Fpn is an electroneutral H+/Fe2+ antiporter in which the transport of each Fe2+ is coupled to the transport of two H+ in the opposite direction.[8] The essential role of FPN1 in dietary iron absorption is supported by the phenotype of mice with FPN1 gene deletion, showing the rapid onset of an anemic condition.[9] A gradient of Fpn as well as of DMT1 expression is observed in the duodenum, with the highest levels observed in the proximal duodenum and the lowest levels in the distal duodenum (Figure 2).[10]
The regulation of intestinal iron absorption requires the coordinated control of three distinct pathways: the hepcidin/ferroportin axis; the IRE/IRP regulatory system; and the hypoxia-regulated HIF system. Hepcidin is a peptide hormone synthesized by liver cells that plays a central and key role in the control of iron metabolism; hepcidin binds to ferroportin and induces its internalization and consequent degradation, with an inhibition of iron efflux from both enterocytes and macrophages.[11-12] Under conditions of iron excess, hepcidin levels increase and determine a downregulation of ferroportin, in turn responsible for a rise of enterocyte iron levels which in turn determine an enhanced degradation of HIF and inactivation of IRPs; the reduced HIF levels determine a decreased transcription of DMT1, Dcytb, NCOA4 and FPN1, thus resulting in a global gene expression/activity modulation aiming to reduce iron absorption. Genetic mouse models and pharmacologic studies using specific inhibitors support a main role of DMT1 and FPN1 expression under iron deficiency conditions.[13,14] Under conditions of low iron availability, the decrease in hepcidin levels determines a maximal activity of the iron efflux from enterocytes, with a consequent activation of the IRE-IRP system, which in turn determines an increase of DMT1 and Dcytb levels and a decrease of ferritin levels, thus limiting the iron-storing capacity of enterocytes. However, FPN1 mRNA cannot be modulated by IRP because in the duodenum, a Fpt transcript lacking IRE is expressed.[15] In parallel, the low enterocyte iron levels determine a stabilization of HIF2α, which in turn stimulates the transcription of DMT1, Dcytb, NCOA4, and ferroportin, thus contributing to enhancing the rate of intestinal iron absorption and the degradation of ferritin via a process named ferritinophagy. Thus, all these mechanisms of gene expression/activity control together contribute, under conditions of iron deprivation, to increase apical iron absorption, to decrease ferritin synthesis, and to increase iron efflux.
Macrophages exert multiple important roles in iron metabolism, particularly related to iron recycling, in that they recycle large amounts of iron derived from hemoglobin degradation, and iron storage, in that they are able to store the iron that is toxic for other cell types.[16]
Iron recycling in macrophages. Macrophages play a crucial role in regulating heme-iron homeostasis, which is based on two main functions. First, a unique population of macrophages in the bone marrow, acting as nurse cells, participates in forming erythroblastic islands needed for erythropoiesis. They do this by phagocytosing and digesting the nuclei of mature erythroblasts while delivering the iron and heme necessary for hemoglobin synthesis.[17] Second, erythrophagocytic macrophages in the red pulp of the spleen, bone marrow, and liver detect membrane changes in aging RBCs and phagocytize them. This process recycles iron back to erythroid progenitors for heme synthesis and hemoglobin production.[17] Erythrophagocytosis of senescent RBCs takes place in the spleen's red pulp. It involves engulfing senescent RBCs into the phagolysosome, where digestion breaks down hemoglobin. Iron-containing heme is transported into the cytosol through Heme Response Gene 1 (HRG1). Inside the cytosol, iron bound to heme is processed by heme oxygenase-1 (HO-1), releasing Fe2+, biliverdin, and CO.
The iron export mechanism based on the activity of the iron exporter ferroportin is well active in macrophages.[18] When FPN1 expression is upregulated by iron treatment or erythrophagocytosis (iron upregulates FPN1 translation, heme upregulates FPN1 expression), FPN1 expression is markedly enhanced at the plasma membrane of macrophages. FPN1 expression in macrophages is required for macrophage iron release in vivo and modulation of innate immune responses.[18] The release of iron from macrophages after erythrophagocytosis is increased by FPN1 overexpression and is downregulated by hepcidin, which markedly reduces FPN1 protein levels.[19]
Circulating Iron
In normal conditions, transferrin-bound iron represents the main form of iron found in blood. Transferrin (Tf) binds up to two iron molecules and maintains iron in a redox-inert state. About 20-40% of iron-binding sites present in transferrin are normally occupied by iron (coefficient of saturation).The structure of Tf consists of two lobes (N-lobe and C-lobe: each of these lobes can bind and release one iron molecule. Under physiologic conditions, four circulating forms of Tf are observed: diferric Tf with two iron atoms bound, monoferric Tf bound to C-lobe, monoferric Tf bound to N-lobe, and apotransferrin; monoferric Tf is the Tf most frequently observed in serum.[20]
Tf delivers iron to tissues by the ubiquitously expressed transferrin receptor 1 (TfR1). TfR1 is overexpressed in erythroid cells that represent the main cell type involved in the utilization of high amounts of iron required for hemoglobin synthesis.[21] A second receptor able to bind Tf is TfR2. TfR2 is the homolog of TfR1, has a lower affinity than TfR1 for Tf, and, under physiological conditions, does not contribute to iron uptake. The tissue distribution of this receptor is more limited than that of TfR1, being restricted to the hepatic and erythroid cells. TfR2 may function as a sensor of transferrin-bound iron levels in the liver and erythroid cells.[22]
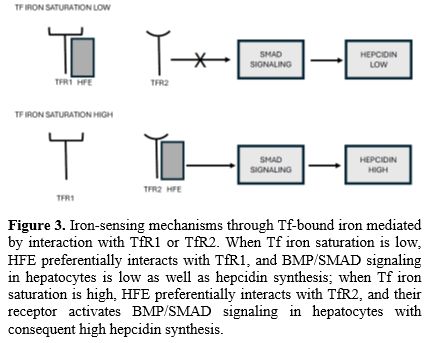
Remarkable differences exist between TfR1 and TfR2 in their capacity to interact with Tf and hereditary hemochromatosis protein HFE: the affinity of TfR2 for holo-Tf is about 25-fold lower than the affinity of TfR1; no detectable binding of HFE to TfR2 was observed, while TfR1 was able to bind HFE protein; in contrast to TfR1, expression of TfR2 is not downmodulated by iron overload, consistent with the lack of IRE in the 3’ untranslated sequence of TfR2 mRNA (Figure 3).[23] However, the membrane levels of both TFR1 and TFR2 are regulated by diferric Tf.
 |
|
HFE (High Ferritin Expression) protein is like the major histocompatibility complex I-type proteins and associates with β2-microglobulin. It is thought that this protein interacts with TfR1 and modulates its activity. Studies in the HeLa cell line showed that HFE overexpression determines a downregulation of iron uptake mediated by TfR1.[24] The interaction between HFE and TfR1 seems to be important for hepcidin regulation in hepatic cells. Mice with knockdown of hepatocyte TfR1 have a moderate decrease of hepatocellular iron, low plasma iron, and inappropriately high hepcidin levels.[25]
In contrast, mice with hepatocellular TfR2 knockout have hepatic iron overload, excess plasma iron, and inappropriately low hepcidin.[26] A more recent study showed that the interaction between HFE and TfR1 is required for the regulation of hepcidin production by hepatic cells.[27] The mutation of HFE is responsible for type I hereditary hemochromatosis.
Uptake and Intracellular Trafficking of Iron in Erythroid Cells
Erythroid cells represent the cells with the highest level of iron uptake, and it was estimated that they contain about 70% of all the iron in the body, incorporated into hemoglobin. Tf-mediated uptake of iron into erythroblasts is regulated by the extent of iron-bound Tf, the number of TfR1, the rate of endocytosis, and exocytosis of TfR1.[3]Developing erythroid cells obtain the iron required for heme and hemoglobin synthesis through binding of diferric Tf mediated by TfR1. The Tf cycle involves three major processes in Tf-TfR1 trafficking: internalization into early endosomes, sorting into recycling endosomes, and recycling to the cell surface. The binding of 2Fe-Tf by TfR1 is followed by endocytosis of the Tf-TfR1 complex; in the acidic environment of the endosome, iron is released from Tf, which remains bound to TfR1. Free ferric iron is then reduced to ferrous iron by STEAP3, a metalloreductase capable of converting iron from insoluble Fe3+ to Fe2+ form that is transported in the cytosol by the metal transporter DMT1 (divalent metal transporter 1). The Tf/TfR1 complex then returns to the cell surface, where apo-Tf dissociates from TfR1.[3,27]
Genetic evidence supports the essential role of TfR1 in normal erythropoiesis; the disruption of the TfR1 gene in mice is embryonic lethal and determines a markedly defective erythropoiesis;[28] TfR1 gene deletion in HSCs determines a markedly defective hematopoiesis with cellular iron deficiency.[29]
Iron taken up by receptor-mediated endocytosis enters a cytosolic labile iron pool (LIP), a pool of chelatable and redox-active iron, which is transitory and serves as a crossroad in iron cell metabolism. A large part of LIP traffics to the mitochondria, where it is utilized for heme synthesis. In erythroid cells, a part of the iron acquired through Tf-mediated endocytosis may be directly transferred to mitochondria through a process of contact between organelles, defined as a kiss-and-run mechanism.[30-31] Alternatively, a part of LIP is transiently captured into ferritin and then released into lysosomes in contact with mitochondria.[32] It is important to note that the extent of LIP is determined by the balance between cellular mechanisms that favor iron uptake, storage, heme degradation, and ferritinophagy.
Heme biosynthesis is controlled by a complex pathway consisting of 8 enzymatic reactions predominantly occurring in mitochondria. Erythroid cells synthesize elevated levels of heme during their differentiation process; in these cells, the rate of heme synthesis is tuned with iron uptake and globin synthesis and is finalized to generate a high level of hemoglobin synthesis. The first enzymatic reaction of the heme biosynthetic pathway consists of the condensation of succinyl-CoA and Glycine to form aminolevulinic acid (ALA). It is catalyzed by the enzyme ALA synthase (ALAS). This enzyme is present in two isoforms, ALAS1 and ALAS2, encoded by two separate genes: ALAS1 is located on chromosome 3 and is ubiquitously expressed, while ALAS2 is located on chromosome X and is exclusively expressed in erythroid tissue. ALAS2 expression strongly increases during late stages of erythroid differentiation, and its deficient expression causes markedly deficient heme and globin synthesis.[33] The expression of ALAS is transcriptionally regulated by the master erythroid transcription factor GATA-1, interacting with an erythroid-specific enhancer, whose loss-of-function mutation is associated with congenital sideroblastic anemia.[34-35] ALAS2 is controlled also at post-transcriptional level by iron through the interaction between IRP (Iron Regulatory Protein) and IRE (Iron Regulatory Element) present in 5’ untranslated region of ALAS2 mRNA: under conditions of iron deficiency, IRPs bind to the 5’ IRE sites of ALAS2 mRNA and inhibit its translation; under conditions of iron abundance, IRPs are either inactivated or degraded and ALAS2 mRNA is translated.[36] There is a very important difference in the regulation of ALAS1 and ALAS2. While ALAS1 activity is negatively regulated by heme levels through a transcriptional repressor via a heme-responsive element, ALAS2 is not negatively regulated by heme.[36] This remarkable difference in ALAS regulation underlines the absolute need to maintain elevated levels of heme synthesis in erythroid cells.
Another important property of erythroid cells consists of the elevated expression and peculiar regulation of ferrochelatase (FECH). The terminal enzyme of the heme biosynthetic pathway catalyzes the conversion of protoporphyrin IX to heme through iron insertion, the rate-limiting enzyme of the heme pathway. The FECH gene is expressed in all tissues to provide heme for essential heme-containing proteins and is upregulated during erythropoiesis for the synthesis of Hb. The human FECH gene promoter contains two GATA and NF-E2 binding sites, which bind their cognate transcription factors and ensure elevated levels of expression of this gene in erythroid cells. In erythroid cells, FECH interacts with different proteins for enzyme stability and with substrates and product transport: FECH forms a complex with mitoferrin-1, a mitochondrial iron transporter and ATP-binding cassette sub-family B member 10 (ABCB 10) to utilize iron imported into mitochondria towards heme biosynthesis; FECH interacts with the erythroid-specific form of ALAS (required for porphyrin precursor production) and with protoporphyrinogen oxidase, the penultimate enzyme in the heme biosynthetic pathway (required for protoporphyrin IX transfer to FECH).[37] These interactions ensure a high rate of heme synthesis in erythroid cells and protect these cells from the accumulation of potentially toxic metabolites, such as protoporphyrin IX and iron.[37] After its synthesis, mitochondrial heme is transferred to hemoproteins and heme-regulated factors, and this process requires chaperoning and trafficking of heme across cellular organelles and compartments. Various iron transporters have been identified. One of these molecules is the mitochondrial and cell surface transporter feline leukemia virus subgroup receptor 1 (FLVCR1). Genetic studies have shown that FLVCR1 is involved in the export of heme out of cells and represents a system to avoid intracellular heme overload; gene inactivation of FLVCR1 in mice causes a block of erythroid cell differentiation at the proerythroblast stage, thus suggesting that coordinate expression of heme and globin is essential for effective erythropoiesis.[38] Two FLVCR1 isoforms exist in erythroid cells. The FLVC1a isoform localizes to the plasma membrane and the FLVCR1b isoform localizes to the mitochondria: the former one is required for the expansion phase of committed erythroid progenitors but cannot drive their terminal differentiation, while the latter one contributes to the expansion phase and is required for erythroid differentiation (Figure 3).[39]
Two different pathways have been suggested to mediate the passage of iron from the cell membrane uptake system to mitochondria: one pathway is ferritin-independent, and the other is ferritin-dependent. The trafficking of iron between various cell compartments requires its binding to a protein that acts as an iron chaperone. PCBP1 acts as a chaperone in erythroid cells, able to deliver iron to human ferritin within erythroid cells; the nuclear receptor coactivator 4 (NCOA4) induces autophagic turnover of ferritin. Genetic studies carried out in mice showed that both the chaperone activity of PCBP1 and the NCOA4-dependent transfer of iron out of ferritin are processes required for efficient iron utilization in erythropoiesis.[39-40] NCOA4 is a cargo receptor that promotes selective autophagy of the iron storage ferritin in conditions of iron deficiency, facilitating iron recovery from cellular stores.[40]
Iron Metabolism Control of Erythropoiesis: The Central Role of Hepcidin/Ferroportin in Systemic Iron Availability
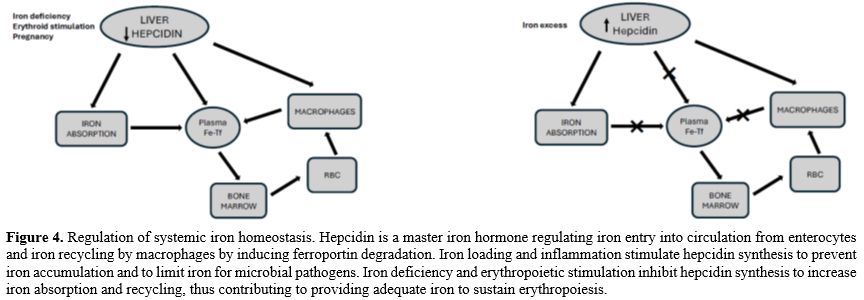
Erythropoiesis is the main consumer of iron in the body. Erythropoietic activity stimulates iron absorption through the modulation of the expression of the gene encoding the iron-regulatory hormone hepcidin, which in turn regulates the release of iron from recycling macrophages and from iron stores in hepatocytes. Increased erythropoiesis is associated with a marked alteration of iron homeostasis to meet the high need for iron to sustain hemoglobin synthesis and erythroid cell proliferation. The coordination between erythropoietic activity and iron homeostasis is mediated by hepcidin, a peptide produced by liver cells, which is a master regulator of systemic iron metabolism. Hepcidin expression is controlled by iron status, erythropoiesis, and inflammation. Hepcidin is a major regulator of body iron balance. Hepcidin controls the rate of iron entry into circulation from intestinal absorption, from recycling macrophages, and hepatocytes through the binding to the iron exporter Ferroportin and inducing its internalization and degradation into lysosomes.[41]At the level of target cells, mainly represented by enterocytes, hepatocytes, and macrophages, hepcidin binds to the iron exporter ferroportin and induces its internalization and degradation, thus inhibiting iron efflux. Hepcidin expression is regulated by iron levels, as well as by the rate of erythropoiesis. Stimulation of erythropoiesis, either mediated by bleeding, hypoxia, or erythropoietin (Epo) administration, markedly inhibits hepcidin expression and, consequently, iron absorption increases during the erythropoietic response to these stimulations to face this increased request for iron. Under these conditions of accelerated erythropoiesis, elevated Epo levels are associated with reduced synthesis of hepcidin. Hepcidin expression is regulated by iron levels: in fact, the transcription of HAMP, Hepcidin Antimicrobial Peptide, the gene encoding hepcidin, is upregulated by iron supply and downregulated by iron deprivation. Stimulation of hepcidin expression inhibits iron absorption from the diet and iron release from recycling macrophages and other iron body stores. In contrast, reducing hepcidin expression promotes iron availability, increasing iron absorption and recycling (Figure 4).[11,42]
 |
|
Bleeding or Epo administration reduced hepcidin synthesis, and that was prevented by suppression of erythropoiesis, thus suggesting that regulation of hepcidin synthesis depends on erythropoietic activity.[43-44] Epo itself could represent the mediator of the effects of erythropoiesis on hepcidin expression. However, studies performed on hepatoma cells and mice lacking EpoR expression in liver cells support the conclusion that hepcidin inhibition mediated by Epo does not require the direct binding of Epo to its liver receptors.[45]
As mentioned above, conditions that resulted in erythropoiesis stimulation, such as Epo administration or phlebotomy, resulted in inhibition of hepcidin expression; importantly, bone marrow ablation prevented the hepcidin inhibition observed in response to phlebotomy or Epo administration.[43,46] These observations suggested that a mediator released by erythroid cells and sensed by liver cells could be responsible for the modulation of hepcidin synthesis exerted by erythropoiesis.
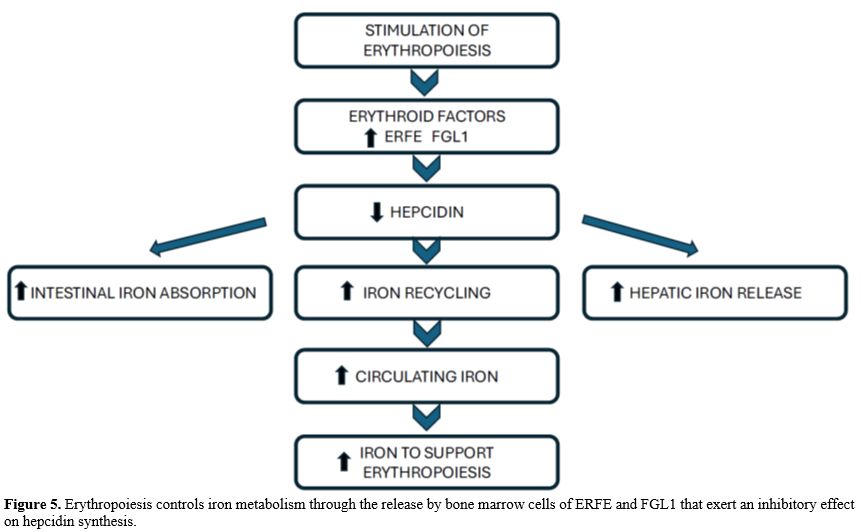
The analysis of bone marrow cells after an acute stimulation of erythropoiesis by phlebotomy led to the identification of the transcript Fam132b mRNA, whose levels markedly increased after phlebotomy (>30 fold 9 hours after phlebotomy); this mRNA encoded the protein called Erythroferrone (ERFE).[47] ERFE expression was also increased after Epo treatment. Erythroid Fam132b mRNA levels mirrored kidney Epo mRNA levels after phlebotomy.[47] Cloning of the gene encoding Fam132b mRNA led to the identification of the Fam132 b gene; this gene encodes a protein initially named FAM132b, but later named ERFE after its role as a hepcidin inhibitor was identified; the ERFE gene encodes in humans a protein of 354 amino acids. ERFE protein is a member of the C1q/TNF-Related Protein (CTPR) family and has a 4-domain structure with a unique N-terminus.[48-49] The mechanism of action of ERFE consists of inhibiting the expression of hepcidin in liver cells, with consequent increased activity of the cellular iron export protein ferroportin and then increased iron absorption from the intestine and mobilization of iron from stores, which can be used to sustain hemoglobin synthesis (Figure 5).[49]
 |
|
The bone morphogenetic protein (BMP)-SMAD signaling is a major transcriptional regulator of hepcidin. Many studies have provided evidence that ERFE inhibits hepcidin production by intersecting with the BMP-SMAD signaling pathway. The presence of SMAD1 or SMAD5 in the liver is essential for maintaining iron homeostasis, while the deficiency of both SMAD1 and SMAD5 causes iron overload.[50] The presence of SMAD1 and SMAD5 is necessary to maintain the response to Epo and ERFE. In fact, both Epo and ERFE failed to suppress hepcidin in mice with a conditional genetic ablation of SMAD1 and SMAD5 in hepatocytes.[50] Subsequent studies have analyzed in more detail the effect of ERFE on the inhibition of BMP-mediated induction of hepcidin. Thus, Arezes and coworkers showed that recombinant ERFE suppresses the BMP/SMAD hepatic pathway; particularly, ERFE specifically abrogated the induction of hepcidin by BMP5, BMP6, and BMP8 but had little or no effect on hepcidin induction by BMP2, BMP4, and BMP9.[51] ERFE binds BMP6 with a greater affinity than BMP2 and BMP4; ERFE interacts with BMP proteins through its N-terminal domain, and this interaction can be inhibited by antibodies against this domain of ERFE.[52]
Srole et al. mapped functionally regions of the ERFE molecule: the hydrophobic helical segment between 81 and 86 and particularly tryptophan residue W82, are essential for interaction with BMP2/BMP6 heterodimer and for ERFE bioactivity; cationic region 96-107 and globular TNFα-like domain 186-354 are required for ERFE multimerization.[53]
ERFE overexpression is expected to induce iron overload, a condition observed in some pathological disorders such as β-thalassemia and other disorders characterized by ineffective erythropoiesis.
To examine the impact of ERFE overexpression, Coffey et al. created transgenic mouse lines with varying levels of ERFE overexpression in erythroid cells; as expected, ERFE-transgenic mice develop relative hepcidin deficiency and iron overload, with severity depending on the level of ERFE overexpression.[53] ERFE-overexpressing mice, especially those with high levels, also show other effects at the liver and systemic cell level.[54]
Interestingly, a recent study showed that ERFE is also expressed in osteoblasts, where its synthesis is stimulated by Epo and hypoxia.[54] Selective gene ablation of ERFE in osteoblasts showed that this ERFE contributes to red cell regeneration in response to stress erythropoiesis.[55]
Studies carried out in normal and anemic rats showed that ERFE levels after Epo administration exhibited a two-wave increase, with an early peak occurring 2 hours and a later peak occurring 8 hours after Epo administration.[56] The early ERFE peak predicted Hb response and Epo responsiveness.[56]
Serum ERFE levels were explored in a large population, including normal individuals and some individuals with anemia. In normal individuals, ERFE, serum ferritin, hepcidin, and soluble TfR (sTfR) levels in males and females of different ages were explored: in males, ferritin, hepcidin, and ERFE levels moderately increased with age, while sTfR levels remained almost constant with some fluctuations; in females, ferritin and hepcidin levels markedly increased after 40 years of age, associated with a significant rise of ERFE levels and sTfR levels almost constant with some fluctuations.[57] ERFE levels were significantly higher in anemic subjects and inversely correlated with Hb, Tf iron saturation, ferritin, and hepcidin levels, and directly correlated with sTfR levels.[56] Finally, high ERFE levels were associated with a significant reduction of hepcidin only in patients with high sTfR levels, reflecting a condition of iron deficiency.[57] The tuning of hepcidin expression according to iron levels implies the capacity of liver cells to sense iron levels. In this context, hepcidin synthesis is controlled by extracellular Tf-bound iron through a mechanism involving TfR1 and TfR2 and by intracellular liver iron content through a mechanism involving BMP (Bone Morphogenetic Protein)/SMAD signaling.
ERFE also plays an important role in fetal erythropoiesis. In conditions of iron-deficient pregnancy, ERFE plays an important role, favoring the redistribution of iron within maternal/fetal compartments in the embryo to support embryonic/fetal erythropoiesis.
However, ERFE plays a minor role during iron-replete pregnancy.
As discussed earlier, some observations suggest that additional factors beyond ERFE may influence hepcidin production after erythropoietic stress. Although ERFE is critical for suppressing hepcidin within the first four hours after such stress, ERFE-deficient mice still recover from anemia caused by hemorrhage and chronic inflammation.[47] Moreover, in thalassemic mice, removing or neutralizing ERFE increases hepcidin levels and reduces Systemic overload; however, simply restoring hepcidin does not completely resolve iron overload. Supporting this idea, a recent study identified hepatokine fibrinogen-like-1 (FGL1) as a hepcidin suppressor that is induced in the liver during hypoxia in the recovery phase after anemia.[58] Similar to ERFE, FGL1 works by binding to BMP6, thereby inhibiting the BMP-SMAD signaling pathway.[58]
Iron-mediated control of erythropoiesis: Role of Transferrin 2
Several studies suggest that TfR2 may act as a mediator of iron's regulation of erythropoiesis. Its ability to sense iron is linked to its interaction with differric Tf, leading to modulation of erythropoiesis by affecting Epo sensitivity in erythroid cells. However, these findings are complex, and it remains unclear how TfR2 influences Epo sensitivity. In vitro studies on TfR2-deficient erythroid cells have shown conflicting results. For example, Forejtnikova and colleagues demonstrated that erythroid progenitors from TfR2-/- mice exhibit decreased Epo sensitivity and elevated circulating Epo levels; in human erythroid progenitors, TfR2 knockdown delays terminal erythroid differentiation and maturation.[59] Conversely, Fouquet et al., using UT7 cells, found that TfR2 knockdown increased EpoR levels due to receptor stabilization, resulting in heightened Epo sensitivity as indicated by increased signaling through ERK, AKT, and STAT5 pathways. The discrepancy between these studies may be due to differences in the cellular systems.[60]Several studies on TfR2 gene knockout supported that TfR2 loss can cause an increased Epo sensitivity of erythroid cells. Thus, Nai et al. have developed a TfR2 bone marrow knockout mouse (TfR2BMKO) by transplanting bone marrow cells from TfR2-/- mice into WT recipients; control chimeric mice were transplanted with bone marrow from WT donors.[22] TfR2BMKO mice displayed increased RBC counts and Hb content, reduced mean corpuscular volume of RBCs within 2-4 months after bone marrow transplantation.[22] These responses were associated with increased erythroid maturation and normal Epo levels. Interestingly, under conditions of mild dietary iron deprivation, determines an increase of erythroblast number, reduces apoptosis and enhances Epo levels in controls, but not in TfR2(BM/KO) mice; under marked dietary iron restriction, erythropoiesis of TfR2BMKO mice was not further modified, as well as Epo levels and EpoR downregulation occurring during erythroid cell differentiation was delayed.[22] These findings were interpreted as supporting an enhanced sensitivity of TfR2-deficient erythroid cells to Epo. In a subsequent study, these authors showed that deletion of bone marrow TfR2 ameliorates anemia and iron overload in a murine model of transfusion-independent beta-thalassemia.[61] These studies have originated the development of a model on the role of TfR2 in the control of systemic iron metabolism and erythropoiesis: in an iron-replete condition, stabilized TfR2 induces iron signaling to hepcidin in the liver and impairs Epo sensitivity in erythroid cells; in iron-deficient conditions, TfR2 destabilization prevents iron signaling to hepcidin in the liver and allows enhanced Epo sensitivity in erythroid cells.
Wortham et al. analyzed erythropoiesis in TfR2-/- mice, showing that: BM erythroid populations of TFR2-/- mice displayed decreased BFU-E, CFU-E, and proerythroblasts and increased orthochromatic erythroblasts, while the spleen of TfR2-/- mice had an increase in both erythroid progenitor proliferation and terminal stages of erythroid differentiation.[62] Importantly, these changes in BM and spleen persisted when liver TfR2 expression was restored in TfR2-deficient mice.[62]
In vitro studies have shown that, in addition to its contribution to an iron sensing mechanism, erythroid TfR2 may also contribute to iron uptake through a mechanism involving transferrin internalization and lysosomal delivery in erythroid progenitors; erythroid mitochondria specifically associate with lysosomes and are regulated by TfR2.[63] A second in vitro study reported the definition of a cellular iron-regulated vesicle transport pathway showing the link between surface TfR2 and EpoR on erythroid progenitors. The main factors of this pathway involve several steps: (i) TfR2 undergoes lysosomal catabolism induced by iron deprivation and this effect is blocked by isocitrate; (ii) TfR2 surface trafficking requires the activity of iron-dependent aconitase enzymes; (iii) TfR2 binds Scribble, a master regulator of receptor trafficking and signaling, and mediates its lysosomal catabolism with iron deprivation, and isocitrate blocks this effect; (iv) Scribble interacts with EpoR and promotes its surface delivery; (v) Scribble downregulation decreases surface EpoR expression, as observed in iron deprivation.[64] Therefore, this pathway involves an upstream sensor represented by TfR2, an intermediary transducing element represented by xxxxx, and an effector target represented by EpoR.[64]
Other studies showed the existence of an iron-dependent mechanism regulating membrane expression of TfR2: cultured human erythroid cells release a soluble form of TfR2, a phenomenon enhanced by iron deprivation and inhibited by diferric transferrin.[65]
Finally, a recent study using conditional knockout of TfR2 in mice strains engineered to express either Nblocked or Cblocked transferrin provided evidence that TfR2 expressed on erythroid cells coordinates iron availability and EpoR sensitivity by only one specific isoform of monoferric forms of Tf.[66]
The ensemble of these results suggests the existence of a mutual crosstalk between iron and erythropoiesis: on one side, erythropoiesis modulates the rate of iron absorption through ERFE; on the other side, iron modulates erythropoiesis through the iron sensor TfR2 which modulates Epo sensitivity of erythroid cells: when is decreased in iron deficiency the Epo sensitivity is increased because TfR2 is removed from the surface of erythroid cells.[67]
Regulation of Cellular Iron Homeostasis: Role of IRE/IRP
As systemic iron availability is adjusted to body iron needs chiefly by the hepcidin-ferroportin (FPN) axis, intracellular iron content is regulated by the Iron Regulatory Element-Iron Regulatory Protein (IRE-IRP) system.[3]This system is based on the presence of IREs (Iron Regulatory Element), short conserved stem-loops, recognized by IRPs (Iron Regulatory Protein). The IREs are found in the untranslated regions of various mRNAs whose encoded proteins are involved in iron metabolism. The IRE/IRP system is orchestrated by the interaction of IRP1 (also known as ACO1) and IRP2 (also known as IREB2) with cis-regulatory IREs present in the untranslated regions (UTR) of mRNAs encoding iron metabolism molecules (Figure 6). When iron level is low, IRPs bind to the 5’UTR IRE of mRNAs encoding the ferritin-H and ferritin-L iron storage proteins, the iron exporter FPN1, ALAS2 or the hypoxia-inducible factor 2 (HIF2A) and inhibit their translation (Figure 6). IRP binding to 3’-UTR IREs in the TfR1 mRNA or DMT1 mRNA protect the transcripts from nuclease-mediated degradation by Regnase-1 and Roquin-1 molecules[68-69] (Figure 6). In iron-rich cells and oxygen-replete cells, IRP2 is displaced from IRE RNA by F-box and leucine-rich repeat protein 5 (FBXL); it is targeted for degradation by proteasome-mediated proteolysis[70] and IRP1 assembles an iron-sulfur cluster and switches its biologic activity to an aconitase.[71]
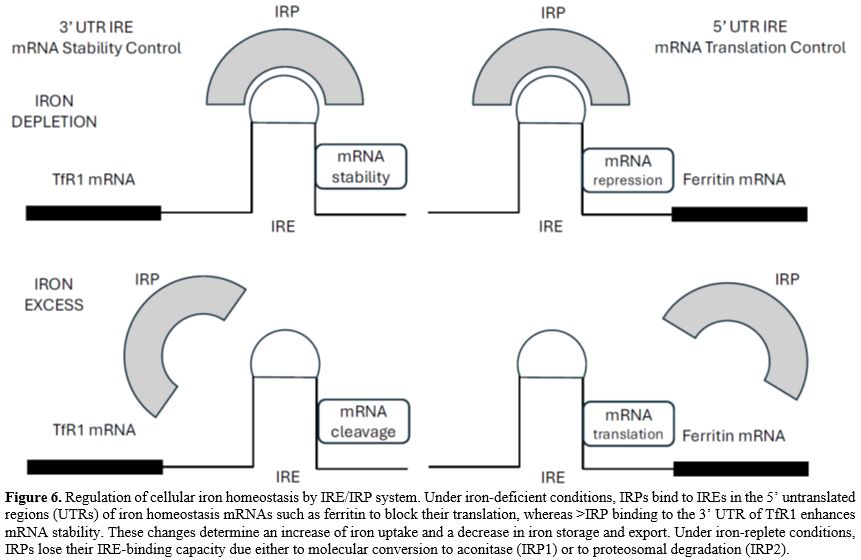 |
|
Genetic studies have supported the necessity of a correct function of IRPs for normal iron homeostasis. Defects in iron-sulfur cluster biogenesis determine abnormal stimulation of IRP activity, with impairment of heme synthesis in erythroid cells;[72] aberrant gain of function of IRP2 function in FBXL5-null mice was found to be lethal.[73] Constitutive systemic IRP2 gene deletion in mice resulted in the development of microcytic anemia, diabetes, and neurologic symptoms.[74-75] IRP1 deficiency determines a transient polycythemia during early life, related to derepression of HIF2α translation in kidney cells and subsequent stimulation of Epo expression.[76] In contrast to single IRP gene knockout studies, double IRP1 and IRP2 gene inactivation studies resulted in early embryonic lethality, thus supporting the absolute necessity of the IRE/IRP pathway and the capacity of one IRP to compensate in part for the loss of the other IRP.[74]
The difference observed between the phenotype induced by IRP1 and IRP2 gene deletion can be explained by the peculiarity of the IRE present in HIF2α mRNA that binds with higher affinity to IRP1 than to IRP2, a finding that helps to explain why IRP1-/- mice exhibit elevated Epo levels and erythrocytosis, while IRP2-/- mice experience a refractory anemia.[77] Furthermore, IRP2-/- cells misregulate iron metabolism when cultured in 3-6% oxygen, a physiological tissue concentration, but not in 21% oxygen, a concentration that activated IRP1.[78] Thus, IRP2 is determinant in the regulation of iron metabolism because it registers iron concentrations and modulates its RNA-binding capacity at physiological oxygen concentrations.
HIF and iron metabolism. Another mechanism of regulation of iron metabolism is dependent upon the hypoxia-inducible factor (HIF) system. Under conditions of low iron and oxygen availability, the regulatory alpha subunits (HIF-1α, HIF-2α, and HIF-3α) form a biologically active complex with subunit beta (HIF-1β or ARNT) and translocate to the nucleus, where they target several genes involved in iron homeostasis and erythropoiesis.[79-80] (Figure 7). Under iron- and oxygen-replete conditions, HIFα subunits are targeted for proteolytic degradation mediated by the oxygen and iron-dependent prolyl-hydroxylases.[79]
A fundamental function of HIF-2α consists of regulating the production of Epo by a group of interstitial fibroblasts in the kidney (Norm cells) that secrete Epo; these cells sense O2 levels and increase the production of Epo when they feel a decrease in O2 availability.[81] When O2 is abundant, HIFs are degraded. Whereas when O2 is low, HIFs accumulate, activate, bind to the Epo gene, and increase the rate at which it is transcribed into mRNA, leading to increased serum Epo levels.
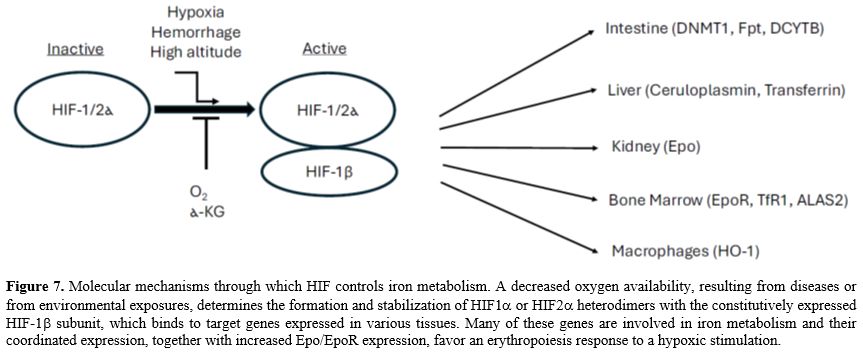 |
|
The other two important targets of HIF-2α are represented by DMT1[82] and ferroportin[83] in duodenal enterocytes, where their expression is upregulated by HIF-2α under iron deficiency conditions. On the other hand, HIF-2α itself is a target of IRP, which controls its translation.[84] HIF activation is modulated by intracellular iron levels also through another mechanism, related to hydroxylase activation, requiring iron as a cofactor.
HIF activation determines an inhibition of hepcidin synthesis by liver cells, but this effect is indirect, being mediated by stimulation of Epo-induced erythropoiesis.[85] Furthermore, hypoxia also induces the synthesis in liver cells of the hepatokine fibrinogen-like 1 (FGL1), a suppressor of hepcidin synthesis.[58,86] Therefore, under hypoxic conditions, there is a coordinated series of events involving the kidney, the liver, and the bone marrow. The kidney increases the production of Epo, while the liver upregulates the serine protease TMPRSS6 to decrease BMP-SMAD signaling and hepcidin synthesis by liver cells. At the level of bone marrow, Epo signaling activation induces an increased production of ERFE peaking at 24h after the hypoxic stimulation, while the liver increases the synthesis and release of FGL1, peaking at 24-72 hours after hypoxic stimulation; both ERFE and FGL1 act inhibiting BMP-SMAD signaling and increasing hepcidin production (peaking at 6-7 days after hypoxic stimulation).[86]
Iron Metabolism During Infections and Inflammation: Role of Hepcidin
Hepcidin synthesis increases during infections and inflammatory conditions, thus suggesting a possible role of hepcidin as a mediator of innate immunity. Hepcidin synthesis in the liver is increased by interleukin-6 (IL-6), a cytokine involved in inflammatory and immune responses; IL-6 acts through the STAT3 signaling pathway and synergistically cooperates with BMP signaling in the transcriptional activation of HMAP gene expression.[87-88] Studies in normal volunteers infused with bacterial lipopolysaccharide showed that elevated hepcidin occurs early during the inflammatory response and is responsible for hypoferremia that develops early during an acute inflammatory response.[89]Studies in murine experimental models of bacterial infections suggest that the inflammatory hepcidin response observed in infectious processes may mediate the prevention of the generation of non-transferrin-bound iron (NTBI) in a condition in which the risk of iron overloading is high due to the suppression of erythropoiesis.[90-91] Iron sequestration into macrophages and hypoferremia due to inflammation-induced hepcidin significantly limit the availability of iron for erythropoiesis and contribute to anemia associated with chronic inflammatory disease.[92]
It is noteworthy that hepcidin synthesis is suppressed during pregnancy; this suppression is likely linked to the production of a yet-unknown placental hormone. This physiological suppression fulfills the need to supply large amounts of iron necessary for fetal growth, erythropoiesis, and the expansion of maternal erythroid cell production. Studies in mouse models have shown that suppressing maternal hepcidin was crucial for ensuring enough iron transfer to the fetus, and the increase in maternal red blood cell mass, along with a placental-derived hepcidin suppressor, appears to play a key role in this physiological adaptation.[93-94] Other factors that help reduce hepcidin production during pregnancy include decreased maternal iron stores from increased utilization and heightened erythropoietic activity. Additionally, the protease TMPRSS6, which inhibits BMP signaling and hepcidin expression, is also involved in the suppression of hepcidin during pregnancy.
Conclusions
In the past four decades, tremendous progress in our understanding of iron metabolism has been made. In this context, the molecular and functional characterization of a set of molecules involved in iron uptake, intracellular iron transport, export out of the cells, transport in circulation, recycling, and intracellular utilization allowed us to define a complex network of iron-related genes. This network needs fine regulation at both cellular and systemic levels. On the other hand, the discovery of hepcidin as a central key regulator of iron metabolism and of hypoxia-regulated pathways allowed a better understanding of the systemic control of iron metabolism through the coordination of iron absorption, recycling, and utilization by erythroid cells.The mechanisms underlying the high uptake of iron by erythroid cells to sustain high heme and hemoglobin synthesis have been in part elucidated and have contributed to understanding the unique properties of these cells and their regulatory role in the context of iron metabolism.
Acknowledge
We thank Dr. Laura Silvestri for her useful advice and suggestions.Abbreviations used in this article
 |
|
References
- Comprubi E, Jordan SF, Vasiliadou R, Lane N. Iron catalysis at the origin of life. IUBMB Life 2017; 69: 373-381. https://doi.org/10.1002/iub.1632 PMid:28470848
- Mleczko-Sanecka K, Silvestri L. Cell-type-specific insights into iron regulatory processes. Am J Hematol. 2021 Jan;96(1):110-127. https://doi.org/10.1002/ajh.26001
- Ganz T. Systemic iron homeostasis. Physiol Rev 2013; 93: 1721-1741. https://doi.org/10.1152/physrev.00008.2013 PMid:24137020
- Correnti,
M.; Gammella, E.; Cairo, G.; Recalcati, S. Iron absorption: molecular
and pathophysiological aspects. Metabolites 2024; 14, 228. https://doi.org/10.3390/metabo14040228 PMid:38668356 PMCid:PMC11052485
- Gunshin,
H.; Fujiwara, Y.; Custodio, A.O.; Direnzo, C.; Robine, S.; Andrews,
N.C. Slc11a2 is required form intestinal iron absorption and
erythropoiesis but dispensable in placenta and liver. J Clin Invest
2005; 115, 1258-1266. https://doi.org/10.1172/JCI200524356 PMid:15849611 PMCid:PMC1077176
- Heoda
J, Shah A, Zhang L. Heme, an essential nutrient from dietary proteins,
critically impacts diverse physiological and pathological processes.
Nutrients 2014; 6: 1080-1102. https://doi.org/10.3390/nu6031080 PMid:24633395 PMCid:PMC3967179
- Kenny
TC, Khan A, Son Y, Yue L, Heissel S, Sharma A, Pasolli HA, Liu Y,
Gamazon ER, Alwaseem H, et al. Integrative genetic analysis identifies
FLVCR1 as a plasma-membrane choline transporter in mammals. Cell Metab
2023; 35, 1057-1071. https://doi.org/10.1016/j.cmet.2023.04.003 PMid:37100056 PMCid:PMC10367582
- Pan,
Y.; Ron, Z.; Gao, S.; Shen, J.; Wang, L.; Xu, Z.; Yu, Y.; Bachina, P.;
Zhang, H.; Pan, X.; et al. Structural basis of iron transport and
inhibition in ferroportin. Nat Commun 2020, 11, 5686. https://doi.org/10.1038/s41467-020-19458-6 PMid:33173040 PMCid:PMC7655804
- Donovan
AS.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.;
Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for
iron homeostasis. Cell Metab 2005, 1, 191-200. https://doi.org/10.1016/j.cmet.2005.01.003 PMid:16054062
- Balusikova,
K.; Dustalikova-Cimburova, M.; Tachechi, I.; Kovar, J. Expression
profiles of iron transport molecules along the duodenum. J Cell Mol Med
2022, 26, 2995-3004. https://doi.org/10.1111/jcmm.17313 PMid:35445529 PMCid:PMC9097835
- Nemeth, E.; Ganz, T. Hepcidin and iron in health and disease. Ann Rev Med 2023, 74, 261-277. https://doi.org/10.1146/annurev-med-043021-032816 PMid:35905974 PMCid:PMC9943683
- Drakesmith, H.; Nemeth, E.; Ganz, T. Ironing out ferroportin. Cell Metab 2015, 22, 777-787. https://doi.org/10.1016/j.cmet.2015.09.006 PMid:26437604 PMCid:PMC4635047
- Mastrogiannaki
M, Matak P, Keith B, Simon C, Vauloint S, Peyssonnaux C. HIF-2,
promotes iron absorption in mice. J Clin Invest 2019, 119, 1159-1166. https://doi.org/10.1172/JCI38499 PMid:19352007 PMCid:PMC2673882
- Schwartz
A, Das NK, Ramakrishnan S, Jain C, Jurkovitch MT, Mu J, Nemeth E,
Lakhal-Littleton S, Colacino JA, Shah YM. Hepatic hepcidin/intestinal HIF-2α axis maintains iron absorption during iron deficiency and overload. J Clin Invest 2019,; 129, 336-348. https://doi.org/10.1172/JCI122359 PMid:30352047 PMCid:PMC6307944
- Zhang,
D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A
ferroportin transcript that lacks an iron-responsive element enables
duodenal and erythroid precursor cells to evade translational
repression. Cell Metab 20'09, 9, 461-473. https://doi.org/10.1016/j.cmet.2009.03.006 PMid:19416716 PMCid:PMC2685206
- Recalcati, S.; Cairo, G. Macrophages and iron: a special relationship. Biomedicines 2024, 9, 1585. https://doi.org/10.3390/biomedicines9111585 PMid:34829813 PMCid:PMC8615895
- Korolnek, T.; Hamza, I. Macrophages and iron trafficking at the birth and death of red cells. Blood 2015, 125, 2893-2897. https://doi.org/10.1182/blood-2014-12-567776 PMid:25778532 PMCid:PMC4424413
- Zhang
Z, Zhang F, An P, Guo X, Shen Y, Tao Y, Wu Q, Zhang Y, Yu Y, Ning B, et
al. Ferroportin 1 deficiency in mouse macrophages impairs iron
homeostasis and inflammatory responses. Blood 2011; 118: 1912-1922. https://doi.org/10.1182/blood-2011-01-330324 PMid:21705499
- Knutson
MD, Oukka M, Koss LM, Aydemir F, Wessling-Resnick M. Iron release from
macrophages after erythrophagocytosis is up-regulated by ferroportin1
overexpression and down-regulated by hepcidin. Proc Natl Acad Sci USA
2005; 102: 1324-1328. https://doi.org/10.1073/pnas.0409409102 PMid:15665091 PMCid:PMC547844
- DSi
Rumo SC, Check IJ, Hunter RL. Quantitation of apo-, mono-, and
differric transferrin by polyacrylamide gradient electrophoresis in
patients with disorders of iron metabolism. Blood 1985; 66: 1445-1451. https://doi.org/10.1182/blood.V66.6.1445.1445
- Sposi
NM, Cianetti L, Tritarelli E, Pelosi E, Militi S, Barberi T,
Gabbianelli M, Saulle E, Kuhn L, Peschle C, Testa U. Mechanisms of
differential transferrin receptor expression in normal hematopoiesis.
Eur J Biochem 2000; 267: 6762-6774. https://doi.org/10.1046/j.1432-1033.2000.01769.x PMid:11082186
- Nai
A, Lindonnici MR, Rausa M. The second transferrin receptor regulates
red blood cell production in mice. Blood 2015; 125: 1170-1179. https://doi.org/10.1182/blood-2014-08-596254 PMid:25499454 PMCid:PMC4399753
- West
AP, Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ.
Comparison of the interactions of transferrin receptor 1 and
transferrin receptor 2 with transferrin and the hereditary
hemochromatosis HFE. J Biol Chem 2000; 275: 38135-38138. https://doi.org/10.1074/jbc.C000664200 PMid:11027676
- Riedel
HD, Muckenthaler MU, Gehrke SG, Mohr I, Brennan K, Herrmann T, Fitscher
BA, Hentze MW, Stremmel W. HFE downregulates iron uptake from
transferrin and induces iron-regulatory protein activity in stably
transfected cells. Blood 1999; 94: 3915-3921. https://doi.org/10.1182/blood.V94.11.3915 PMid:10572108
- Fillebeen
C, Charlebois E, Wagner J, Katsarou A, Mui J, Vali H, Garcia-Santos D,
Ponka P, Presle J, Pantopoulos K. Transferrin receptor 1 controls
systemic iron homeostasis by fine-tuning hepcidin expression to
hepatocellular iron load. Blood 2019; 133: 344-355. https://doi.org/10.1182/blood-2018-05-850404 PMid:30538134
- Wallace
DF, Summerville L, Subramaniam VN, Targeted disruption of the hepatic
transferrin receptor 2 gene in mice leads to iron overload.
Gastroenterology 2007; 132: 301-310. https://doi.org/10.1053/j.gastro.2006.11.028 PMid:17241880
- Xiao
X, Moschetta GA, Xu Y, Fisher AL, Alfaro-Magallanes VM, Dev S, Wang CY,
Babitt JL. Regulation of iron homeostasis by hepatocyte TfR1 requires
HFE and contributes to hepcidin suppression in -thalassemia. Blood 2023; 141: 422-431. https://doi.org/10.1182/blood.2022017811 PMid:36322932 PMCid:PMC9936306
- Levy
JE, Jin D, Fujiwara Y, Kuo F, Andrews NC. Transferrin receptor is
necessary for development of erythrocytes and nervous system. Nat Genet
1999; 21: 396-399. https://doi.org/10.1038/7727 PMid:10192390
- Wang
S, He X, Wu Q, Jiang L, Chen L, Yu Y, Zhang P, Huang X, Wang J, et al.
Transferrin receptor 1-mediated iron uptake plays an essential role in
hematopoiesis. Haematologica 2020; 105: 2071-2082. https://doi.org/10.3324/haematol.2019.224899 PMid:31601687 PMCid:PMC7395265
- Das
A, Nag S, Mason AB, Barroso MM. Endosome-mitochondria interactions are
modulated by iron release from transferrin. J Cell Biol 2016; 214:
831-845. https://doi.org/10.1083/jcb.201602069 PMid:27646275 PMCid:PMC5037410
- Hamdi
A, Roshan TM, Kahawita TM, Mason AB, Sheftel AD, Ponka P. Erythroid
cell mitochondria receive endosomal iron by a "kiss-and-run" mechanism.
Biochim Biophys Acta 2016; 1863: 2859-2867. https://doi.org/10.1016/j.bbamcr.2016.09.008 PMid:27627839
- Aronova
MA, Noh SJ, Zhang G, Byrne C, Meier ER, Kim YC, Meier ER, Leapman R.
User of dual-electron probes reveals the role of ferritin as an iron
depot in ex vivo erythropoiesis. iScience 2021; 24: 102901. https://doi.org/10.1016/j.isci.2021.102901 PMid:34401678 PMCid:PMC8355919
- Harigae
H, Suwabe N, Weinsock PH, Nagai M, Fiujita H, Yamamoto M. Deficient
heme and globin synthesis in embryonic stem cells lacking the
erythroid-specific delta-aminolevulinate synthase gene. Blood 1998; 91:
798-805. https://doi.org/10.1182/blood.V91.3.798 PMid:9446639
- Kaneko
K, Furuyama K, Fujiwara T, Kobayashi R, Ishida H, Hariagae H.
Identification of the novel erythroid-specific enhancer for ALAS2 gene
and its loss-of-function mutation associated with congenital
sideroblastic anemia. Haematologica 2014; 99: 252-261. https://doi.org/10.3324/haematol.2013.085449 PMid:23935018 PMCid:PMC3912954
- Surinya
KH, Cox TC, May BK. Identification and characterization of a conserved
erythroid-specific enhancer located in intron 8 of the human
5-aminolevulinate synthase 2 gene. J Biol Chem 1998; 273: 16798-16809. https://doi.org/10.1074/jbc.273.27.16798 PMid:9642238
- Kramer
MF; Gunaratne P, Ferreira GC. Transcriptional regulation of the
erythroid-specific 5-aminolevulinate synthase gene. Gene 2000; 247:
153-166. https://doi.org/10.1016/S0378-1119(00)00103-7 PMid:10773455
- Obi
CD, Dailey HA, Jam-Alahmedi Y, Wohlschgel JA, Medlock AE. Proteome
analysis of ferrochelatase interactome in erythroid and non-erythroid
cells. Life 2023; 13: 577. https://doi.org/10.3390/life13020577 PMid:36836934 PMCid:PMC9958551
- Doty
RT, Phelps RT, Shadle C, Sanchez-Bonilla M, Kael SB, Abkowitz JL.
Coordinate expression of heme and globin is essential for effective
erythropoiesis. J Clin Invest 2015; 125: 4681-4691. https://doi.org/10.1172/JCI83054 PMid:26551679 PMCid:PMC4665774
- Mercurio
S, Petrillo S, Chiabrando D, Bassi ZI, Gays D, Camporeale A, Vacaru A,
Miniscalco B, Valperga G, Silengo L, et al. The heme exporter FLVCR1
regulates expansion and differentiation of committed erythroid
progenitors by controlling intracellular heme accumulation.
Haematologica 2015; 100: 720-729. https://doi.org/10.3324/haematol.2014.114488 PMid:25795718 PMCid:PMC4450617
- Ryu
MS, Zhang D, Protchenko O, Shakavry-Elizeh M, Phipott CC. PCBP1 and
NCO4 regulate erythroid iron storage and heme biosynthesis. J Clin
Invest 2017; 127: 1786-1797. https://doi.org/10.1172/JCI90519 PMid:28375153 PMCid:PMC5409075
- Ji
X, Jha A, Humenik J, Ghanem LR, Kromer A, Duncan-Lewis C, Tarxler E,
Weiss MJ, Barash Y, Leibhader SA. RNA-binding proteins PCBP1 and PCBP2
are critical determinants of murine erythropoiesis. Mol Cell Biol 2021;
41: e00668-20. https://doi.org/10.1128/MCB.00668-20 PMid:34180713 PMCid:PMC8384066
- Nemeth
E, Tuttle MS, Powelson J. Hepcidin regulates cellular iron efflux by
binding to ferroportin and inducing its internalization. Science 2004;
306: 2090-2093. https://doi.org/10.1126/science.1104742
- Pak
M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin
during anemia requires erythropoietic activity. Blood 2006; 108:
3730-3735. https://doi.org/10.1182/blood-2006-06-028787 PMid:16882706 PMCid:PMC1895477
- Sasaki
Y, Noguchi-Sasaki M, Yasuno H, Yorozu K, Shimonaka Y. Erythropoietin
stimulation decreases hepcidin expression through hematopoietic
activity on bone marrow cells in mice. Int J Hematol 2012; 96: 692-700.
https://doi.org/10.1007/s12185-012-1217-4 PMid:23160767
- Gammella
E, Diaz V, Recalcati S, Buratti P, Sanoya M, Day S, Noguchi C, Gasmann
M, Cairo G. Erythropoietin,s inhibiting impact on hepcidin expression
occurs indirectly. Am J Physiol Integr Com Physiol 2015; 308:
R330-R335. https://doi.org/10.1152/ajpregu.00410.2014 PMid:25519735 PMCid:PMC4347750
- Vokurka
M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver
respond to inhibition of erythropoiesis. Physiol Rev 2006; 55: 667-674.
https://doi.org/10.33549/physiolres.930841 PMid:16497104
- Kautz
I, Jung G, Valore EV, Rivella S, Nometh E, Ganz T. Identification of
erythroferrone as an erythroid regulator of iron metabolism. Nat Genet
2014; 46: 678-684. https://doi.org/10.1038/ng.2996 PMid:24880340 PMCid:PMC4104984
- Srole
DN, Ganz T. Erythroferrone structure, functions, and physiology: iron
homeostasis and beyond. J Cell Physiol 2021; 236: 4888-4901. https://doi.org/10.1002/jcp.30247 PMid:33372284 PMCid:PMC8026552
- Kautz L, Nemeth E. Molecular liaisons between erythropoiesis and iron metabolism. Blood 2014; 124: 479-482. https://doi.org/10.1182/blood-2014-05-516252 PMid:24876565 PMCid:PMC4110655
- Wang
CYY, Core AB, Canali S, Zumbrennen-Bullough KB, Ozer S, Umans L,
Zwijsen A, Babill JL. Smad 1/5 is required for erythropoietin-mediated
suppression of hepcidin in mice. Blood 2017; 130: 73-83. https://doi.org/10.1182/blood-2016-12-759423 PMid:28438754 PMCid:PMC5501149
- Azeres
J, Foy N, McHigh K, Sawant A, Quinker D, Terraube V, Brinth A, Tam M,
LaVallie ER, Taylor S, et al. Erythroferrone inhibits the induction of
hepcidin by BMP6. Blood 2018; 132: 1473-1477. https://doi.org/10.1182/blood-2018-06-857995 PMid:30097509 PMCid:PMC6238155
- Azeres
J, Foy N, McHigh K, Quinkert D, Benard S, Sawant A, Frost JN, Armitage
AE, Pasricha SR, Lim PJ, et al. Antibodies against the erythroferrone
N-terminal domain prevent hepcidin suppression and ameliorate murine
thalassemia. Blood 2020; 135: 547-557. https://doi.org/10.1182/blood.2019003140 PMid:31899794 PMCid:PMC7046598
- Srole
DN, Jung G, Waring AJ, Nemeth E, Ganz T. Characterization of
erythoferrone structural domains relevant to its iron-regulatory
function. J Biol Chem 2023; 299: 105374. https://doi.org/10.1016/j.jbc.2023.105374 PMid:37866631 PMCid:PMC10692919
- Coffey
R, Jung G, Olivera JD, Karin G, Pereira RC, Nemeth E, Ganz T. Erythroid
overproduction of erythroferrone causes iron overload and developmental
abnormalities in mice. Blood 2022; 139: 439-449. https://doi.org/10.1182/blood.2021014054 PMid:34614145 PMCid:PMC8777203
- Phatthalung
PN, Ginburg Y, van Caloen G, Planoutene M, Gumerova AA, Witztum R,
Ingber E, Sultana F, Korkzmaz F, Levy M, et al. Osteoblast-derived
erythroferrone regulates stress erythropoiesis. Blood 2024;
144(suppl.1): 2470. https://doi.org/10.1182/blood-2024-203624
- Xu
P, Wong R, Yan X. Early erythroferrone levels can predict the long-term
haemoglobin responses to erythropoiesis-stimulating agents. Br J
Pharmacol 2024; 1871: 2833-2850. https://doi.org/10.1111/bph.16396 PMid:38653449
- Busti
F, Zidanes AL, Bertolone L, Martinelli N, Castagna A, March G, Vianello
A, Bozzini C, Pramstaller P, Girelli D. Serum erythroferrone levels in
a large population: towards better understanding of connections between
erythropoiesis and homeostasis. Blood 2023; 42(suppl.1): 1096-1097. https://doi.org/10.1182/blood-2023-177943
- Sardo
U, Perrier P, Cormier K, Sotin M, Personnaz J, Medjbeur T, Desquesnes
A, Cannizzo L, Ruiz-Martinez M, Thevenin J, et al. The hepatokine FGL1
regulates hepcidin and iron metabolism during anemia in mice by
antagonizing BMP signaling Blood 2024; 143: 1282-1290. https://doi.org/10.1182/blood.2023022724 PMid:38232308 PMCid:PMC11103088
- Forejtnikova
H, Viellvoey M, Zermati Y, Lambert M, Pellegrino RM, Guihard S, Gaudry
M, Camaschella C, Lacombe C, Roetto A, et al. Transferrin receptor 2 is
a component of the erythropoietic complex and is required for efficient
erythropoiesis. Blood 2010; 116: 5357-5367. https://doi.org/10.1182/blood-2010-04-281360 PMid:20826723
- Fouquet
G, Thongsaad U, Lefevre C, Rousseau A, Tanhaud N, Khongkla E,
Saengsawang W, Anurathapan U, Haugeng S, Maciel TT, et al. Iron-loaded
transferrin potentiates erythropoietin effects on erythroblast
proliferation and survival: a novel role through transferrin receptors.
Exp Hematol 2021; 99: 12-20. https://doi.org/10.1016/j.exphem.2021.05.005 PMid:34077792
- Artuso
I, Liddonici MR, Altamura S, Mandelli G, Pettinato M. Muckenthaler M,
Silvestri L, Ferrari G, Camaschella C, Nai A. Transferrin receptor 2 is
a potential novel therapeutic target for -thalassemia: evidence from a
murine model. Blood 2018; 132: 2287-2296. https://doi.org/10.1182/blood-2018-05-852277 PMid:30209118 PMCid:PMC6302281
- Wortham
AM, Goldman DC, Chen J, Fleming WH, Enns CA. Extrahepatic deficiency of
transferrin receptor 2 is associated with increased erythropoiesis
independent of iron overload. J Biol Chem 2020; 215: 3906-3917. https://doi.org/10.1074/jbc.RA119.010535 PMid:32054685 PMCid:PMC7086028
- Khalil
S, Holy M, Grado S, Fleming R, Kurita R, Nakamura Y, Goldfarb A. A
specialized pathway for erythroid delivery through lysosomal
trafficking of transferrin receptor 2. Blood Adv 2017; 1: 1181-1190. https://doi.org/10.1182/bloodadvances.2016003772 PMid:29296759 PMCid:PMC5728310
- Khalil
S, Delehanty L, Grado S, Holyy M, White III Z, Freeman K, Kurita R,
Nakamura Y, Bullock G, Goldfarb A. Iron modulation of erythropoiesis is
associated with Scribble-mediated control of the erythropoietin
receptor. J Exp Med 2018; 215: 661-679. https://doi.org/10.1084/jem.20170396 PMid:29282252 PMCid:PMC5789406
- Pagani
A, Villevoyye M, NMai A, Rausa M, Ladli M, Lacombe C, Mayeux P, Verdier
F, Camaschella C. Regulation of cell surface transferrin receptor-2 by
iron-dependent cleavage and release of a soluble form. Haematologica
2015; 100: 458-465. https://doi.org/10.3324/haematol.2014.118521 PMid:25637053 PMCid:PMC4380718
- Guerra
A, Sharma P, An X, Parrow NL, Fleming R, Ginzburg Y, Rivella S. TFR2
coordinates erythroid iron availability and erythropoietin receptor
sensitivity by monoferric transferrin forms. Blood 2024; 144(suppl.1):
2468-2469. https://doi.org/10.1182/blood-2024-211984
- Camaschella
C, Pagani A, Silvestri L, Nai A. The mutual crosstalk between iron and
erythropoiesis. Int J Hematol 2022; 116: 182-191. https://doi.org/10.1007/s12185-022-03384-y PMid:35618957
- Yoshinaga
M, Nakatsuka Y, Vandenbon A, Ori D, Uehata T, Tsujimura T, Suzuki Y,
Mino T, Takeuchi O. Regnase-1 maintains iron homeostasis via the
degradation of transferrin receptor 1 and
prolyl-hydroxylase-domain-containing protein 3 mRNAs. Cell Rep 2017;
19: 1614-1630. https://doi.org/10.1016/j.celrep.2017.05.009 PMid:28538180
- Corral
VM, Schultz ER, Eisenstein RS; Connell GJ. Roquin is a major mediator
of iron-regulated changes of transferrin receptor-1 stability. iScience
2021; 24: 102360. https://doi.org/10.1016/j.isci.2021.102360 PMid:33898949 PMCid:PMC8058555
- Wang
H, Shi H, Rajan M, Canarie ER, Hong S, Simoneschi D, Pagano M, Bush MF,
Stoll S, Leibold EA, et al. FBXL5 regulates IRP2 stability in iron
homeostasis via an oxygen-responsive [2FE2S] cluster. Mol Cell 2020;
78: 38-41e5. https://doi.org/10.1016/j.molcel.2020.02.011 PMid:32126207 PMCid:PMC7159994
- Rouault
TA, Maio N. Biogenesis and functions of mammalian iron-sulfur proteins
in the regulation of iron homeostasis and pivotal metabolic pathways. J
Biol Chem 2017; 292: 12744-12753. https://doi.org/10.1074/jbc.R117.789537 PMid:28615439 PMCid:PMC5546015
- Ward DM, Cloonan SM. Mitochondrial iron in human health and disease. Annu Rev physiol 2019; 81: 453-482. https://doi.org/10.1146/annurev-physiol-020518-114742 PMid:30485761 PMCid:PMC6641538
- Moroishi
T, Nishiyama M, Takeda Y, Iwai K, Nakaayma KI. The FXBL5-IRP2 axis is
integral to control of iron metabolism in vivo. Cell Metab 2011; 14:
339-351. https://doi.org/10.1016/j.cmet.2011.07.011 PMid:21907140
- Wilkinson N, Pantopoulos K. The IRP/IRE system in viìvo: insights from mouse models. Front Pharmacol 2014; 5: 176. https://doi.org/10.3389/fphar.2014.00176 PMid:25120486 PMCid:PMC4112806
- De
Santos MCF, Anderson CP, Nechen S, Zumbrennen-Bullough KB, Romney SJ,
Kahle-Stepohan M, Rathkolb B, Gailus-Darner V, Fuchs H, Wolf E, et al.
IRP2 regulates insulin production through iron-mediated
Cdkal1-catalyzed tRNA modification. Nat Commun 2020; 11: 296. https://doi.org/10.1038/s41467-019-14004-5 PMid:31941883 PMCid:PMC6962211
- Wilkinson
N, Pantopoulos K. IRP1 regulates erythropoiesis and systemic iron
homeostasis by controlling HIF2 mRNA translation. Blood 2013; 122:
1658-1668. https://doi.org/10.1182/blood-2013-03-492454 PMid:23777768
- Zhang
DL, Ollivierre H, Qi CF, Rouault TA. A bulge uridine IRE allows IRP1
but not IRP2 to selectively regulate HIF2 expression and ensuing EPO
levels. Blood 2025; 145: 533-542. https://doi.org/10.1182/blood.2024025246 PMid:39316647
- Meyron-Holtz
EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate
iron-regulatory protein activities in vivo. Science 2004; 306:
2087-2090. https://doi.org/10.1126/science.1103786 PMid:15604406
- Peissonnaux C, Nizet V, Johnson RS. Role of the hypoxia inducible factors HIF in iron metabolism. Cell Cycle 2008; 7: 28-32. https://doi.org/10.4161/cc.7.1.5145 PMid:18212530
- Gruber
M, Hu CJ, Johnson RS, Brown EJ, Keith B, Simon MC. Acute postnatal
ablation of Hif-2 results in anemia. Proc Natl Acad Sci USA 2007; 104:
23012-2306. https://doi.org/10.1073/pnas.0608382104 PMid:17284606 PMCid:PMC1892942
- Kragesteen
B, Giladi A, David E, Halevi S, Geisdottir L, Lempke O, Li B, Bapst AM,
Xie K, Katzenelenbogen Y, et al. The transcriptional and regulatory
identity of erythropoietic producing cells. Nat Med 2023; 29:
1191-1200. https://doi.org/10.1038/s41591-023-02314-7 PMid:37106166
- Shah
YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal
hypoxia-inducible transcription factors are essential for iron
absorption following iron deficiency. Cell Metab 2009; 9: 152-164. https://doi.org/10.1016/j.cmet.2008.12.012 PMid:19147412 PMCid:PMC2659630
- Taylor
M, Qu A, Anderson ER. Hypoxia-inducible factor HIF-2 mediates the
adoptive increase of intestinal ferroportin during iron deficiency in
mice. Gastroenterology 2011; 140: 2044-2055. https://doi.org/10.1053/j.gastro.2011.03.007 PMid:21419768 PMCid:PMC3109109
- Anderson
SA, Nizzi CP, Chang YI. The IRP1-HIF-2 axis coordinates iron and
oxygen sensing with erythropoiesis and iron absorption. Cell Metab
2013; 17: 282-290. https://doi.org/10.1016/j.cmet.2013.01.007 PMid:23395174 PMCid:PMC3612289
- Liu
Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates
hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest 2012;
122: 4635-4644. https://doi.org/10.1172/JCI63924 PMid:23114598 PMCid:PMC3533545
- Silvestri L. Ironing erythroid cells takes FGL1 and ERFE to tango. Blood 2024; 143: 1208-1209. https://doi.org/10.1182/blood.2023023645 PMid:38546639
- Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, Ganz T. IL-6 mediates hypoferremia of inflammation by induing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest 2004; 113: 1271-1276. https://doi.org/10.1172/JCI200420945 PMid:15124018 PMCid:PMC398432
- Armitage AE, Eddowes LA, Gileadi U, Cole S, Spottiswood N, Selvakumar TA, Ho LP, Towsend A, Drakesmith H. Hepcidin regulation by immune and infectious stimuli. Blood 2011; 118: 4129-4139. https://doi.org/10.1182/blood-2011-04-351957 PMid:21873546
- Kemna
E, Pickkers P, Nemeth E, van der Hoeven H, Swinkels D. Time-course
analysis of hepcidin, serum iron, and plasma cytokine levels in humans
injected with LPS. Blood 2005; 106: 1864-1866. https://doi.org/10.1182/blood-2005-03-1159 PMid:15886319
- Kim
A, Fung E, Parikh SG, Valore EV, Gabayan V, Nemeth E, Ganz T. A mouse
model of anemia of inflammation: complex pathogenesis with partial
dependence on hepcidin. Blood 2014; 123: 1129-1136. https://doi.org/10.1182/blood-2013-08-521419 PMid:24357728 PMCid:PMC9632791
- Stefanova
D, Raychev A, Arezes J, Ruchala P, Gabayan V, Skurnik M, Dillon BJ,
Horwitz MA, Ganz T, Bulut Y, Nemeth E. Endogenous hepcidin and its
agonist mediate resistance to selected infections by clearing
non-transferrin-bound iron. Blood 2017; 130: 245-257. https://doi.org/10.1182/blood-2017-03-772715 PMid:28465342 PMCid:PMC5520472
- Ganz T. Anemia of inflammation. N Engl J Med 2017; 381: 1148-1157. https://doi.org/10.1056/NEJMra1804281 PMid:31532961
- Sangkhae
V, Ganz T, Nemeth E. Maternal hepcidin suppression is essential for
healthy pregnancy. Blood 2020 136 (suppl.1): 43-44. https://doi.org/10.1182/blood-2020-141148
- Sangkhae
V, Fisher AL, Chua KJ, Ganz T, Nemeth E. Maternal hepcidin determines
embryo iron homeostasis in mice. Blood 2020; 136: 2206-2216. https://doi.org/10.1182/blood.2020005745 PMid:32584957 PMCid:PMC7645983