The clinical reality for aging SCD and thalassemia patients is sobering. Progressive organ damage, chronic pain, and endocrine dysfunction make both disorders profoundly morbid.[3] Yet palliative and hospice services remain criminally underutilized. A 2022 analysis of inpatient data in the U.S. found that less than 0.5% of hospital admissions for SCD patients involved a palliative care consultation, despite the clinical complexity inherent in these diseases.[4] Greek clinicians in a 2023 survey reported limited formal training in end-of-life communication and symptom management, translating into delayed referrals to palliative specialists and fragmented care across the disease trajectory.[5] The consequence is tangible: patients endure prolonged suffering, undergo unnecessary intensive interventions, and die in settings misaligned with their preferences. Why does this happen? Several factors converge: physicians struggle to prognosticate non-malignant diseases; opioid access remains restricted in many regions; cultural beliefs complicate discussions of withdrawal of treatment; and there simply are no disease-specific pathways guiding when and how to refer a thalassemia or SCD patient to hospice.
International hematology and palliative care organizations have begun to acknowledge this gap.[6] Early palliative care - not waiting for terminal decline - can improve pain control, quality of life, symptom management, and patient coping.[6] The evidence now suggests that palliative care should be offered alongside disease-directed therapy, not as a substitute when curative options are exhausted. This narrative review synthesizes literature from 2020 to 2025, international guidelines, and clinical expert consensus to outline how hemoglobinopathy patients can receive comprehensive end-of-life care grounded in clinical evidence, ethical reasoning, and cultural sensitivity. We address clinical manifestations at end-stage, psychosocial support needs, moral dilemmas, and practical referral pathways.
Clinical Manifestations and Disease Progression at End-Stage
SCD and β-thalassemia end-stage presentations could hardly be more different, yet both are devastating (Table 1). SCD is capricious. Patients experience sudden, life-threatening crises punctuated by periods of relative stability, making it nearly impossible to predict when the final decline will come. |
|
In advanced SCD, chronic pain dominates. Unlike acute pain crises, which are episodic, chronic pain from repeated vaso-occlusions-causing avascular necrosis, joint destruction, and non-healing leg ulcers becomes the patient's constant companion. Opioid tolerance develops over years, and patients may require extraordinary doses just to achieve partial relief.[7] Acute chest syndrome, pulmonary hypertension, and cardiac arrhythmias lurk in the background. Silent strokes and overt strokes erode cognition and motor function. Progressive chronic kidney disease, often overlooked until it is severe, compounds morbidity. Hepatic dysfunction emerges from transfusion-related iron overload or hepatitis C acquired from older blood supplies. Functional asplenia renders patients vulnerable to fulminant infections and sepsis.[7-11] These complications rarely announce themselves in an orderly sequence; instead, they accumulate in unpredictable ways, making the prognosis frustratingly elusive.
β-thalassemia is more predictable: a slow, relentless accumulation of iron in the heart and liver, year after year of transfusions and chelation, until cardiac restrictive cardiomyopathy or cirrhosis becomes terminal, but its trajectory is more insidious.[12] Cardiac complications account for major mortality in thalassemia cohorts.[11] Despite three decades of iron chelation therapy, many patients accumulate myocardial iron over decades, resulting in restrictive cardiomyopathy and life-threatening ventricular arrhythmias.[11] Hepatic cirrhosis, particularly in patients with chronic hepatitis C, becomes a major source of morbidity and mortality. Endocrine complications, such as hypogonadism, diabetes, hypothyroidism, and adrenal insufficiency, weaken immune function and physiological reserve. Osteoporosis from iron overload, hypogonadism, and chronic transfusion causes chronic pain and fragility fractures. Splenectomy, performed in some patients for transfusion dependence reduction, paradoxically increases infection risk.[12-15] A recent Greek study tracking 2,475 thalassemia and SCD patients over 12 years found that hepatocellular carcinoma (17.5%), sepsis (11.5%), and heart failure (21.8%) dominated thalassemia mortality; while sepsis (14.9%), hepatic failure (13.9%), and stroke (13.6%) were most common in SCD.[16]
Psychosocial and Spiritual Dimensions of End-of-Life Care
Ask an SCD patient about their suffering, and they will tell you about more than pain. They talk about lost opportunities. The hospital stays that derailed their education. The job they couldn't keep because of unpredictable crises. The relationships that didn't survive the demands of chronic illness. As terminal illness approaches, these accumulated losses crystallize into existential despair. Depression and anxiety are common; post-traumatic stress symptoms emerge in patients who have endured decades of medical crises.[11,17-19] The stigma surrounding pain management, the accusation that they seek opioids for their own purposes rather than for legitimate relief, leaves deep psychological scars and erodes trust in healthcare providers.[2,17-19]For thalassemia patients, the psychological burden is different but equally profound. Lifelong medical dependence, visible body changes from iron overload, and the knowledge that their life expectancy has always been limited create a particular kind of existential weight. Many express guilt about the burden they place on families. Some grieve about the possibility of never having children or raising them. Discrimination in employment and education narrows opportunities, deepening hopelessness.[20,21] Clinical social workers, psychologists, and peer support groups have demonstrated value in reducing isolation and restoring dignity. Spiritual and religious frameworks matter enormously. Some patients find profound meaning in prayer or meditation; others find it in serving their communities. Palliative teams must respect these frameworks and, when possible, facilitate connection with spiritual advisors who understand both their faith traditions and their medical reality.[20,21]
The Council of Europe has explicitly stated that people with life-limiting conditions deserve access to mental health services and family-centered psychosocial support throughout their illness.[22] This is not a luxury; it is a human right. Yet in practice, psychiatrists and psychologists are rarely embedded in hemoglobinopathy centers. Families are often left to navigate existential questions on their own.
Multidisciplinary Palliative Care: Why It Matters
Comprehensive end-of-life care for patients with hemoglobinopathies cannot be delivered by a single physician. SCD demands hematologists who understand the disease, palliative care specialists who excel at symptom control, pain management experts for opioid-tolerant patients, cardiologists, nephrologists, hepatologists, psychiatrists, psychologists, and social workers.[23] β-thalassemia requires similar breadth: cardiologists because cardiac failure dominates; hepatologists because cirrhosis is common; endocrinologists because multiple gland dysfunction is the rule.The critical insight is that palliative care should not replace disease-directed care; it should complement it. A patient approaching end-of-life may still benefit from a transfusion, not to prolong life indefinitely, but to manage a painful complication or allow attendance at a family milestone. This "concurrent care" model, wherein palliative specialists and hematologists work alongside one another, has been shown to improve both symptom control and quality of life.[21,23] It also facilitates earlier, more authentic conversations about goals of care. In Europe, the ERN-EuroBloodNet network has begun promoting multidisciplinary care standards and sharing best practices across specialized centers.[24] Such coordination can reduce disparities and ensure that patients across different healthcare systems receive comparable quality of end-of-life support (Figure 1).
 |
|
Early integration of palliative principles - not late integration - improves pain control, quality of life, coping, symptom self-management, and patient satisfaction.[25,26] Outpatient palliative clinics can initiate these conversations and begin symptom optimization months or years before terminal decline. This approach reduces crisis-driven emergency care and hospitalization. The evidence is strong; the barriers are organizational and cultural.
Ethical and Cultural Complexities at End-of-Life
End-of-life decisions for patients with hemoglobinopathy are never purely medical. Patients from African American, Afro-Caribbean, Middle Eastern, South Asian, and Mediterranean communities bring deep cultural and religious frameworks that shape how they understand illness, treatment, and death. Some patients prefer personal autonomy in decision-making; others defer to family or religious elders. Clinicians must navigate this diversity thoughtfully, as some ethicists call it, "negotiated autonomy", exploring what information patients want, how much control they desire, and what role family should play.[27,28]Emerging gene therapies and HSCT add ethical complexity. When should a desperately ill patient with severe end-organ damage be offered a high-risk curative intervention? Denying it might feel like abandonment; offering it might constitute harm. The answer lies in transparent dialogue grounded in bioethical consultation. The principle of non-maleficence ("do no harm") must be weighed against respect for autonomy and hope.[29-31]
De-escalation or withdrawal of transfusions in thalassemia, or of mechanical ventilation in severe acute chest syndrome, raises profound questions about what constitutes "giving up". In reality, these decisions reflect a compassionate shift from prolonging life to permitting death with dignity and minimal suffering. Yet families shaped by historical trauma and systemic racism often interpret withdrawal of care as rejection. Effective communication demands professional medical interpretation when needed, genuine respect for cultural values, and collaboration with trusted community or religious leaders.[32-34]
The Council of Europe's Resolution 2249 (2018) explicitly recognizes access to pain relief and palliative care as fundamental human rights and calls for the removal of legal barriers to opioid access. In some regions, high-dose opioids raise ethical concerns. Yet, they are ethically defensible under the principle of "double effect" when the intent is symptom control, not hastening death, and when the dose is proportionate to the therapeutic goal. Meticulous documentation of clinical reasoning and dosing protects both patients and clinicians.
Referral Guidelines and Practical Implementation
Prognostication in patients with hemoglobinopathies is notoriously difficult. Unlike cancer, where predictable terminal decline is common, SCD can cause sudden death at nearly any time, and β-thalassemia's decline is gradual but variable. This unpredictability has led to late or missed palliative care referrals. The solution is to base referral criteria not solely on life expectancy estimates, but also on functional status and objective signs of end-organ failure.[16]Specific referral triggers should include: (1) severe cardiomyopathy or pulmonary hypertension requiring continuous oxygen; (2) hepatic failure with synthetic dysfunction; (3) recurrent ICU admissions for vaso-occlusive crises, acute chest syndrome, or sepsis; (4) functional decline (Palliative Performance Scale <50% or ECOG ≥4); (5) cessation of meaningful benefit from disease-modifying therapy; or (6) explicit patient preference for comfort-focused care.[35] When any are present, palliative care consultation should be offered (Figure 2).
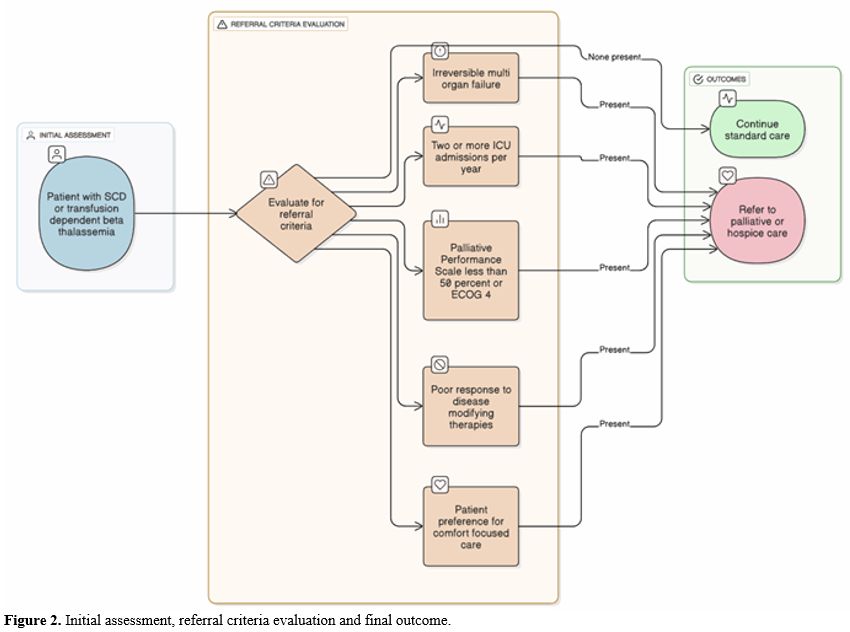 |
|
A major structural barrier exists in the U.S.: Medicare hospice eligibility criteria are cancer-focused and poorly accommodate hemoglobinopathy patients. Physicians must carefully document specific clinical scenarios, e.g., "end-stage SCD with multiorgan dysfunction and intractable pain" or "β-thalassemia with severe cardiac iron overload and ventricular arrhythmias", to justify hospice eligibility. The proposed Hospice CARE Act would permit limited transfusions within hospice, removing a primary barrier to enrollment among transfusion-dependent patients.[36] Current regulations often force an impossible choice: enroll in hospice and forget transfusions or continue transfusions and delay hospice — a false dichotomy that results in late or missed referrals.
Early integration of outpatient palliative clinics is the antidote to abrupt, crisis-driven transitions. These clinics can support symptom management and advance care planning months in advance, while concurrent care models permit disease-modifying therapies to continue when used primarily for symptom relief.[37,38] Such integration bridges hematologic and palliative philosophies and promotes genuine long-term shared decision-making.
Structured, culturally sensitive conversations, especially during periods of clinical stability, should precede major deterioration. Discussions should occur at transitions (pediatric to adult care), after sentinel events (stroke, heart failure), and periodically thereafter. These repeated conversations align interventions with evolving priorities: returning home comfortably, attending important family events, or minimizing procedures. Data from England in 2022 reveal that only 4.7% of hemoglobinopathy patients died in hospice, while 56.6% died at home, suggesting systemic underutilization or inaccessibility of structured palliative services.[39] Institutions should track outcomes, place of death, adequacy of pain control, bereavement satisfaction, and implement quality improvements when gaps appear.
Countries including Italy, France, and Greece have pioneered integrated care models with multidisciplinary clinics, seamless transitions from pediatric to adult care, and embedded psychosocial support. These models deserve replication and study. A persistent obstacle is limited access to transfusions in home-hospice settings. Emerging evidence supports flexible hospice models that permit palliative transfusions when they improve comfort, without artificially prolonging life.[40] As disease-specific hospice guidelines evolve, core principles are clear: early referral, functional and clinical criteria (not just life expectancy), flexibility in protocols, and genuinely holistic, culturally grounded care ensure dignified, patient-centered end-of-life experiences.
Conclusions and Future Directions
Sickle cell disease and transfusion-dependent β-thalassemia are no longer death sentences in childhood; they are chronic illnesses that demand lifelong engagement with healthcare systems and carry substantial morbidity even with optimal disease-modifying therapy. As these patients age and approach end-of-life, they merit the same high-quality, anticipatory, multidisciplinary, and culturally informed palliative care that we have come to expect for cancer patients. Yet the evidence shows they do not receive it. Palliative services remain fragmented, referrals are delayed or absent, opioid access is restricted, and few healthcare systems have established disease-specific end-of-life pathways. This is not an insurmountable problem but a care-delivery gap that can be closed through deliberate health system redesign. Healthcare organizations must develop explicit, disease-specific referral criteria and pathways for patients with hemoglobinopathies. Flexible hospice models should be implemented, permitting palliative transfusions and other disease-specific therapies when they meaningfully improve comfort. Equitable access to opioids and other analgesics must be ensured, particularly in resource-limited regions where opioid availability remains catastrophically low. Psychosocial and spiritual support must be systematically embedded within hemoglobinopathy centers rather than added as an afterthought. Hematology trainees require formal education in end-of-life communication, symptom management for opioid-tolerant patients, and cultural humility. Finally, patients and families need disease-specific education about palliative care and hospice, correcting persistent misconceptions that these services equate to abandonment. The evidence is available; the clinical pathway is clear. What remains is the will to implement it.Author Contributions
All authors made substantial contributions to the conception and design of the review, the acquisition, analysis, and interpretation of the literature, and the drafting and critical revision of the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work, ensuring its accuracy and integrity.References
- Weatherall DJ. The challenge of haemoglobinopathies
in resource-poor countries. British journal of haematology. 2011
Sep;154(6):736-44. https://doi.org/10.1111/j.1365-2141.2011.08742.x PMid:21726207
- Makani
J, Cox SE, Soka D, Komba AN, Oruo J, Mwamtemi H, Magesa P, Rwezaula S,
Meda E, Mgaya J, Lowe B. Mortality in sickle cell anemia in Africa: a
prospective cohort study in Tanzania. PloS one. 2011 Feb 16;6(2):e14699
https://doi.org/10.1371/journal.pone.0014699 PMid:21358818 PMCid:PMC3040170
- Farmakis
D, Porter J, Taher A, Cappellini MD, Angastiniotis M, Eleftheriou A.
2021 Thalassaemia International Federation guidelines for the
management of transfusion-dependent thalassemia. Hemasphere. 2022 Aug
1;6(8):e732. https://doi.org/10.1097/HS9.0000000000000732 PMid:35928543 PMCid:PMC9345633
- Nwogu-Onyemkpa
E, Dongarwar D, Salihu HM, Akpati L, Marroquin M, Abadom M, Naik AD.
Inpatient palliative care use by patients with sickle cell disease: a
retrospective cross-sectional study. BMJ open. 2022 Aug
1;12(8):e057361. https://doi.org/10.1136/bmjopen-2021-057361 PMid:35973707 PMCid:PMC9386219
- Konstantis
A, Paraskeva M, Avgoustidi M, Kiagia M, Kounnis V, Rovithi M, Alexaki
M, Ardavanis A, Arvanitou E, Tsoukalas N, Agelaki S. The implementation
of palliative care in Greece: a position paper by the Palliative Care
Working Group of the Hellenic Society of Medical Oncology (HeSMO). In
Forum of Clinical Oncology 2025 Jan 31 (Vol. 15, No. 1, pp. 4-13). https://doi.org/10.2478/fco-2023-0038
- Ahmadi
M, Rassouli M, Gheibizadeh M, Ebadi A, Asadizaker M. Experiences of
Iranian Patients with Thalassemia Major Regarding Their Palliative and
Supportive Care Needs: A Qualitative Content Analysis. International
Journal of Community Based Nursing and Midwifery. 2025 Apr 1;13(2):113
- Ballas
SK, Kesen MR, Goldberg MF, Lutty GA, Dampier C, Osunkwo I, Wang WC,
Hoppe C, Hagar W, Darbari DS, Malik P. Beyond the definitions of the
phenotypic complications of sickle cell disease: an update on
management. The Scientific World Journal. 2012;2012(1):949535. https://doi.org/10.1100/2012/949535 PMid:22924029 PMCid:PMC3415156
- Piel FB, Steinberg MH, Rees DC. Sickle cell disease. New England Journal of Medicine. 2017 Apr 20;376(16):1561-73. https://doi.org/10.1056/NEJMra1510865 PMid:28423290
- Yawn
BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH,
Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ. Management
of sickle cell disease: summary of the 2014 evidence-based report by
expert panel members. Jama. 2014 Sep 10;312(10):1033-48. https://doi.org/10.1001/jama.2014.10517 PMid:25203083
- Lanzkron
S, Carroll CP, Haywood Jr C. Mortality rates and age at death from
sickle cell disease: US, 1979-2005. Public health reports. 2013
Mar;128(2):110-6. https://doi.org/10.1177/003335491312800206 PMid:23450875 PMCid:PMC3560868
- Cannas
G., Poutrel S., Virot E., Marie M., Guilhem A., El-Kanouni A., Bourgeay
R., Mutumwa M.G., Elhamri M., Hot A.Health-related quality of life
measurement in adults with sickle cell disease in steady state:
Experience of one French reference center.Mediterr J Hematol Infect Dis
2025, 17(1): e2025065. https://doi.org/10.4084/MJHID.2025.065 PMid:40937312 PMCid:PMC12422244
- Taher AT, Musallam KM, Cappellini MD. β-Thalassemias. New England Journal of Medicine. 2021 Feb 25;384(8):727-43. https://doi.org/10.1056/NEJMra2021838 PMid:33626255
- Ganz T, Nemeth E. Pathogenic mechanisms in thalassemia II: iron overload. Hematology/Oncology Clinics. 2023 Apr 1;37(2):353-63. https://doi.org/10.1016/j.hoc.2022.12.006 PMid:36907608
- Carsote
M, Vasiliu C, Trandafir AI, Albu SE, Dumitrascu MC, Popa A, Mehedintu
C, Petca RC, Petca A, Sandru F. New entity-thalassemic endocrine
disease: major beta-thalassemia and endocrine involvement. Diagnostics.
2022 Aug 9;12(8):1921. https://doi.org/10.3390/diagnostics12081921 PMid:36010271 PMCid:PMC9406368
- Chiew
JY, Thiruchelvam J, Rahmat MB, William SP, Shafien ZB, Banerjee KG. The
key complications of beta thalassemia major: a review and update. Int J
Res Med Sci. 2021 Jun;9:1846-52. https://doi.org/10.18203/2320-6012.ijrms20212263
- Delicou
S, Manganas K, Diamantidis MD, Venou TM, Delaporta P, Pantelidou D,
Spachiou E, Tsagia S, Pappi V, Petropoulou F, Kapsali E. Comparative
analysis of mortality patterns and treatment strategies in thalassaemia
and sickle cell disease patients: A 12-year study. British Journal of
Haematology. 2025 May;206(5):1466-78 https://doi.org/10.1111/bjh.20043 PMid:40090899
- Essien
EA, Winter-Eteng BF, Onukogu CU, Nkangha DD, Daniel FM. Psychosocial
challenges of persons with sickle cell anemia: A narrative review.
Medicine. 2023 Nov 24;102(47):e36147. https://doi.org/10.1097/MD.0000000000036147 PMid:38013366 PMCid:PMC10681612
- Raphael
JL. Addressing social determinants of health in sickle cell disease:
the role of Medicaid policy. Pediatric blood & cancer. 2020
May;67(5):e28202. https://doi.org/10.1002/pbc.28202 PMid:32037648
- Treadwell
MJ, Anie KA. Quality of life in sickle cell disease: what matters.
Hematology/Oncology Clinics. 2022 Dec 1;36(6):1137-49. https://doi.org/10.1016/j.hoc.2022.06.010 PMid:36400535
- Forni
GL, Grazzini G, Boudreaux J, Agostini V, Omert L. Global burden and
unmet needs in the treatment of transfusion-dependent β-thalassemia.
Frontiers in Hematology. 2023 Jun 20;2:1187681. https://doi.org/10.3389/frhem.2023.1187681
- Bizri
M, Koleilat R, Akiki N, Dergham R, Mihailescu AM, Bou-Fakhredin R,
Musallam KM, Taher AT. Quality of life, mood disorders, and cognitive
impairment in adults with β-thalassemia. Blood Reviews. 2024 May
1;65:101181. https://doi.org/10.1016/j.blre.2024.101181 PMid:38341336
- Council
of Europe (PACE). Resolution 2249 (2018) on the right to palliative
care. Strasbourg: Parliamentary Assembly of the Council of Europe;
2018. Available from: https://pace.coe.int/en/files/24982/html
- Patel
RV, Nwogu-Onyemkpa E, Kanter J, Osunkwo I. Palliative Care for Adults
With Sickle Cell Disease: An Unrecognized Opportunity. The
Hematologist. 2024 Sep 1;21(5). https://doi.org/10.1182/hem.V21.5.2024514
- European Commission. European Reference Networks. Available from: https://health.ec.europa.eu/rare-diseases/european-reference-networks_en
- Schatz
AA, Oliver TK, Swarm RA, Paice JA, Darbari DS, Dowell D, Meghani SH,
Winckworth-Prejsnar K, Bruera E, Plovnick RM, Richardson L. Bridging
the gap among clinical practice guidelines for pain management in
cancer and sickle cell disease. Journal of the National Comprehensive
Cancer Network. 2020 Apr 1;18(4):392-9.
https://doi.org/10.6004/jnccn.2019.7379 PMid:32259777 PMCid:PMC9764961
- Erondu
MU, Boateng S. Equity in Palliative Care: Interviews with Leaders of a
Sickle Cell Community-Facing Organization. Journal of Pain and Symptom
Management. 2024 May 1;67(5):e644-5. https://doi.org/10.1016/j.jpainsymman.2024.02.088
- Eleftheriou
A, Forni GL, Sayani F. Multidisciplinary Care and Reference Centres in
addressing haemoglobin disorders. Guidelines for the Management of
Transfusion-Dependent β-Thalassaemia (TDT)[Internet]. 5th edition. 2025.
- Anie KA. The intersection of sickle cell disease, stigma, and pain in Africa. Hematology. 2024 Dec 6;2024(1):240-5. https://doi.org/10.1182/hematology.2024000549 PMid:39644047 PMCid:PMC11665707
- Berghs
M, Ebenso B, Ola B. Social Determinants of Severity in Sickle Cell
Disease. In Sickle Cell Disease in Sub-Saharan Africa 2024 Apr 30 (pp.
17-30). Routledge. https://doi.org/10.4324/9781003467748-3
- Munung
NS, Nnodu OE, Moru PO, Kalu AA, Impouma B, Treadwell MJ, Wonkam A.
Looking ahead: ethical and social challenges of somatic gene therapy
for sickle cell disease in Africa. Gene Therapy. 2024 May;31(5):202-8. https://doi.org/10.1038/s41434-023-00429-7 PMid:38012299 PMCid:PMC11090833
- van
Hooff LC, Merz EM, Kidane Gebremeskel AS, de Jong JA, Burchell GL,
Lunshof JE. Balancing benefits and burdens: a systematic review on
ethical and social dimensions of gene and cell therapies for hereditary
blood diseases. BMC medical ethics. 2025 Mar 14;26(1):36. https://doi.org/10.1186/s12910-025-01188-3 PMid:40087738 PMCid:PMC11907911
- Darby
JE, Akpotu IC, Wi D, Ahmed S, Doorenbos AZ, Lofton S. A scoping review
of social determinants of health and pain outcomes in sickle cell
disease. Pain Management Nursing. 2025 Feb 1;26(1):e1-9. https://doi.org/10.1016/j.pmn.2024.09.002 PMid:39370347
- Cáceres-Titos
MJ, Porras-Santana JM, Cabillas-Romero MR, Begoña García-Navarro E.
Managing cultural diversity in end-of-life care: a qualitative study.
BMC palliative care. 2025 May 4;24(1):124. https://doi.org/10.1186/s12904-025-01759-6 PMid:40320533 PMCid:PMC12049785
- Asamoah
EH. Perceptions and Experiences of Sickle Cell Disease Patients and
Parents/Caretakers on Alternative Treatment Options for Pain Management
(Doctoral dissertation, Walden University).
- Benjamin
LJ. Pain management in sickle cell disease: palliative care begins at
birth? Hematology Am Soc Hematol Educ Program. 2008:466-474.
https://ashpublications.org/hematology/article/2008/1/466/19906/Pain-management-in-sickle-cell-disease
https://doi.org/10.1182/asheducation-2008.1.466 PMid:19074128
- Kayastha
N, LeBlanc TW. Palliative care for patients with hematologic
malignancies: are we meeting patients' needs early enough? Expert
Review of Hematology. 2022 Sep 2;15(9):813-20. https://doi.org/10.1080/17474086.2022.2121696 PMid:36062508
- Shaw,
PA A. Hospice Criteria: Determining That Time Is Limited. In the Arc of
Conversation: A How-to Guide for Goals of Care Conversations 2025 Feb
14 (pp. 91-157). Cham: Springer Nature Switzerland. https://doi.org/10.1007/978-3-031-70495-6_5
- Reyes,
J., & Lin, J. (2025). Bridging Pediatric and Adult Palliative Care
to Amplify Interdisciplinary Home Hospice Support. Journal of Pain and
Symptom Management, 69(5), e585-e586. https://doi.org/10.1016/j.jpainsymman.2025.02.250
- Office for National Statistics. Deaths registered in England and Wales: 2022. Newport, UK: ONS; 2023. Available from
https://www.ons.gov.uk/peoplepopulationandcommunity/birthsdeathsandmarriages/deaths/datasets/deathsregisteredinenglandandwalesreferencetables
- Brinkman-Stoppelenburg
A, Rietjens JA, Van der Heide A. The effects of advance care planning
on end-of-life care: a systematic review. Palliative medicine. 2014
Sep;28(8):1000-25. https://doi.org/10.1177/0269216314526272 PMid:24651708