Case Description
A 40-year-old man was diagnosed with acute myeloid leukaemia (AML) with CEBPα monoallelic mutation in 2001, for which he was treated with an intensive induction chemotherapy and subsequent allogeneic transplant from a 10/10 unrelated donor. The post-transplant follow-up was unremarkable. Twenty years after the transplant, he showed up in our outpatient transplant clinic reporting a new onset of headache, visual disturbance, and hearing loss. His symptoms rapidly worsened, and the blood exams were significant for thrombocytopenia and mild leukopenia. We performed a lumbar puncture, and cerebrospinal fluid analysis showed 80% myeloblasts; the bone marrow aspirate showed 70% myeloblasts. We analysed hematopoietic chimerism, which was molecularly evaluated using Variable Number of Tandem Repeats (VNTR) analysis of bone marrow mononuclear cells and purified peripheral blood T lymphocytes, both immunomagnetically selected. Bone marrow analysis showed 66% donor chimerism, whereas chimerism analysis of CD3+ T cells in peripheral blood was full donor; collectively, these data suggested disease relapse. The myeloblast phenotype was positive for CD34, CD117, CD13, and CD33, and cytogenetic and next-generation sequencing (NGS) analysis showed that leukaemia was the original disease, characterised by a CEBPα monoallelic mutation, excluding donor-derived or therapy-related AML. Given the rapidly worsening neurological clinical presentation, we decided to immediately proceed with a course of chemotherapy with methotrexate and high-dose cytarabine. We performed tumour lysis syndrome prophylaxis with rasburicase 0.2 mg/kg (15 mg daily) for three days.On the first day of therapy, after the infusion of 15 mg of rasburicase, 3500 mg/sqm of methotrexate, and a single dose of cytarabine (2000 mg/sqm), we observed an abrupt arterial oxygen desaturation (80%), with the patient being completely asymptomatic and eupnoic. Arterial oxygen saturation (SO2) levels were not affected by high-flow oxygen, and a chest CT scan was negative for pulmonary embolism. We performed an arterial blood gas test, which showed normal pH and blood gas values, but also methemoglobinemia (MetHb) at 8.5%. Given high levels of MetHb, we hypothesized an enzymatic defect in red blood cells, particularly a deficiency of glucose-6-phosphate dehydrogenase (G6PD). We checked the G6PDH level in peripheral blood, which was markedly reduced (15 mU/10^9 RBC; range 220-570). As part of the differential diagnosis of anemia with high levels of MetHb, we checked the blood levels of NADH-cytochrome b5 reductase, whose deficiency is associated with a rare genetic form of methemoglobinemia, which were normal. Twenty-four hours later, the patient experienced an acute haemolytic anemia. As other potential differential diagnoses, Coombs test was negative, excluding a warm autoimmune hemolitic anemia; the patients was afebrile, with normal levels of serum lactate and C-reactive protein, excluding a sepsis-related event; rasburicase was the only drug related to a possible drug-induced hemolysis; methotrexate and cytarabine are not known as potential cause of drug-induced hemolitic anemia and the patient did not take other oral medicaments. Pre-hemolysis hemoglobin levels were 10.2 gr/dL, LDH 391 U/L (normal value up to 240 U/L), total bilirubin 5.8 mg/dl (direct bilirubin 0.4 mg/dL) and normal values of haptoglobin; during the peak of the hemolysis, hemoglobin levels was 7.1 gr/dl, LDH 1380 U/L, total bilirubin 9,5 mg/dL and haptoglobin was undosable; trends in haemolytic parameters during the hemolitic crisis are reported in Figure 1. The clinical picture, together with the results of the enzymatic test, confirmed our hypothesis of an acute haemolytic crisis due to rasburicase administration in a patient with previously unknown G6PD deficiency. MetHb levels were below 30%, which is the recommended threshold for administering methylene blue; therefore, we provided only supportive care for the acute haemolytic anaemia.
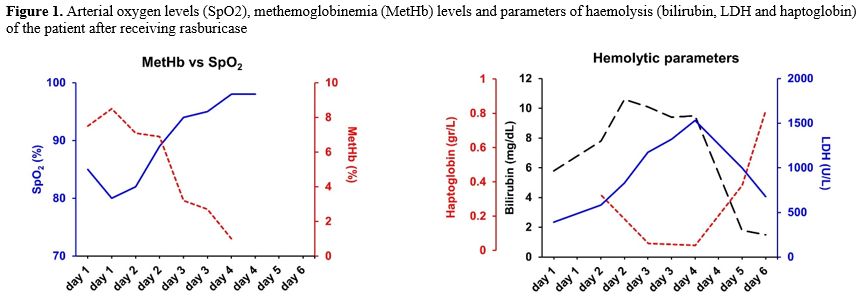 |
|
The chronological events of this clinical case are summarized in Figure 2. Since the patient's personal history was negative for red blood cell disorders and he never experienced such hemolytic events before the first allogeneic transplant, which could have prompted the suspicion of G6PD deficiency, we checked the donor's geographical origin. We discovered that he is from Sardinia, an area with a high prevalence of G6PD deficiency. This corroborated our hypothesis that the G6PDH deficiency was donor-derived. We reported this event to the Italian Bone Marrow Donor Registry (IBMDR), but the donor was lost to follow-up. Our patient fully recovered from the hemolytic event, achieved a hematological remission, and then underwent a second allogeneic transplant from a different unrelated donor. During his follow-up, we checked the G6PDH blood levels three months after transplant, which turned out to be normal.
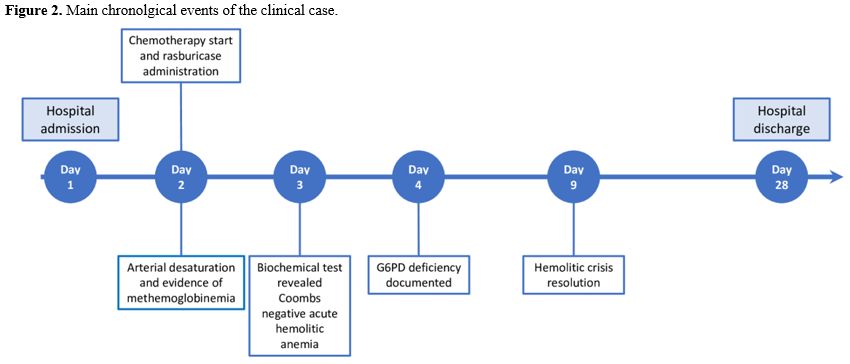 |
|
Discussion
Three points of relevance from this case should be highlighted:1) Late relapse of AML twenty years after allogeneic transplant is very rare; among different casuistries, it happens in 1-3% of all relapses[1-3] and it commonly occurs as extramedullary disease, such as central nervous system (CNS) involvement, which suggests a loss of immunological control of the donor's immune system, given the fact that the CNS is a sanctuary site for immune system.[4,5] The cytogenetic and NGS analysis ruled out a secondary AML (therapy-related AML) and allowed us to identify the exact same disease as twenty years ago. The chimerism analysis documented a full donor lymphoid chimerism and loss of myeloid chimerism, consistent with relapse of the original disease. This clinical case highlights the importance of biobanking cells at diagnosis to run additional tests in case of late relapse, not only for diagnostic purposes but also for translational research.
2) Rasburicase is associated with the rise of methaemoglobin, which is characterized by oxidation of Fe2+ to Fe3+, reducing the ability of haemoglobin to release oxygen to tissues. Under physiological conditions, reductions to Fe2+ are guaranteed by two biochemical pathways, one NADH-dependent (through cytochrome b5 reductase) and the other NADPH-dependent. Methemoglobinemia and hemolysis are linked to rasburicase because its mechanism of action implies the synthesis of hydrogen peroxide during the conversion of uric acid to allantoin. Thus, rasburicase should be avoided in patients with G6PD deficiency, as it may cause an acute haemolytic crisis and methaemoglobinemia due to the increased sensitivity of red blood cells to oxidative stress. There are a few reports in the literature concerning rasburicase-induced hemolytic crisis in patients with hematological malignancies.[6-10] Concerning hematopoietic stem cell transplantation, there is a small case series from China in which five patients with known G6PD deficiency were transplanted from G6PD-deficient donors; the Author did not find significant differences in red cell engraftment or the incidence of hemolytic events.[11] In this regard, a medical history of G6PD deficiency should always be investigated before rasburicase treatment. However, there are no reported cases of donor-derived G6PD deficiency, unlike the case in our patient. We diagnosed the G6PD deficiency in our patient by measuring the serum levels of the enzyme; given the impossibility to check the donor’s G6PD levels and the mixed hematopoietic chimerism in the bone marrow, a possible confirm of the donor origin of the deficiency would have been the same test on patient’s erytroid and myeloid progenitors, which should have been normal on the patient’s cells and defective in donor’s cells.
3) Current IBMDR (Italian Bone Marrow Donor Registry) guidelines exclude from donation patients known for enzymatic disease of the red blood cell; likewise, screening for potential donors coming from regions endemic for G6PDH deficiency is not required. In this regard, Pilo et al. reported a prevalence of G6PD deficiency of 19% among 101 donors from Sardinia, a region with a high prevalence. In this work, G6PDH deficiency did not result in a risk factor for donors in terms of haemolytic crisis during myelostimulation, nor affected the quality of stem cell harvest, and, with a 10-year follow-up, no differences were observed among recipients in terms of erythroid recovery or haemolytic events.[12] Even though stem cell mobilization and collection appear to be safe in donors with G6PD deficiency, the possible occurrence of post-engraftment hemolytic events, similar to our patient, poses the question of a possible role of active screening for donors coming from regions endemic for G6PD deficiency, especially if the donor has a personal or familial history of anemia or hemolytic events. In such cases, we suggest an alternative donor or, if the identified donor is the sole available and the transplant is unamendable, we would properly instruct the recipient to avoid specific drugs (i.e., cotrimoxazole). The role of an active screening program for red blood cell enzymatic defects in potential stem cell donors, particularly for those from high-prevalence geographic areas, could be an interesting topic for discussion among International Donor Registries, taking into account ethical and financial considerations.
Funding
We thank the Associazione Italiana per la Ricerca sul Cancro-AIRC 5 x 1000 Program and the Associazione Italiana Lotta alla Leucemia, Linfoma e Mieloma (AIL) Sezione Bergamo.Authors contribution
GC, FL, MF, MCM, AA and AG treated the patient; OS performed NGS analysis; CP designed figures; AR revised the paper and provided major intellectual contribution; All Authors revised the manuscript and gave the final approval before submission.Patient’s consent statement
The patient signed a privacy informed consent which was approved by the Institutional Review Board of our Hospital.References
- Mariani, S. et al. Very late acute myeloid leukemia
relapse: clinical features, treatment and outcome. Leukemia and
Lymphoma vol. 62 1022-1025 Preprint at https://doi.org/10.1080/10428194.2020.1713320 PMid:31942824
- Medeiros,
B. et al. Characteristics and outcomes of acute myelogenous leukemia
patients with very late relapse (>5 years). Leuk Lymphoma 48, 65-71
(2007). https://doi.org/10.1080/10428190601043252 PMid:17325849
- Verma,
D. et al. Late relapses in acute myeloid leukemia: Analysis of
characteristics and outcome. Leuk Lymphoma 51, 778-782 (2010). https://doi.org/10.3109/10428191003661852 PMid:20196624 PMCid:PMC4086357
- Clark,
W. B., Strickland, S. A., Barrett, A. J. & Savani, B. N.
Extramedullary relapses after allogeneic stem cell transplantation for
acute myeloid leukemia and myelodysplastic syndrome. Haematologica vol.
95 860-863 Preprint at https://doi.org/10.3324/haematol.2010.025890 PMid:20513805 PMCid:PMC2878780
- Chong,
G., Byrnes, G., Szer, J. & Grigg, A. Extramedullary Relapse after
Allogeneic Bone Marrow Transplantation for Haematological Malignancy.
Bone Marrow Transplantation vol. 26 www.nature.com/bmt (2000). https://doi.org/10.1038/sj.bmt.1702659 PMid:11100282
- Marinacci,
L. X., Simeone, F. J., Lundquist, A. L., Kuter, D. J. & Mahowald,
G. K. Case 38-2020: A 52-Year-Old Man with Cancer and Acute Hypoxemia.
New England Journal of Medicine 383, 2372-2383 (2020). https://doi.org/10.1056/NEJMcpc2004991 PMid:33296564
- Hammami,
M. B. et al. Rasburicase-induced hemolytic anemia and
methemoglobinemia: a systematic review of current reports. Annals of
Hematology vol. 103 3399-3411 Preprint at https://doi.org/10.1007/s00277-023-05364-6 PMid:37468669
- Wilson,
J. Rasburicase-induced methaemoglobinaemia and catastrophic oxidative
haemolysis in undiagnosed G6PD deficiency. British Journal of
Haematology vol. 200 7 Preprint at https://doi.org/10.1111/bjh.18464
(2023). https://doi.org/10.1111/bjh.18464 PMid:36120836
- Sonbol,
M. B., Yadav, H., Vaidya, R., Rana, V. & Witzig, T. E.
Methemoglobinemia and hemolysis in a patient with G6PD deficiency
treated with rasburicase. Am J Hematol 88, 152-154 (2013). https://doi.org/10.1002/ajh.23182 PMid:22573495
- Ahmed, M., Sanchez, T., Norgbe, S., Picking, C. R. & Millner, P. G. Rasburicase-Induced Methemoglobinemia. Cureus https://doi.org/10.7759/cureus.14406
- Au,
W. Y. et al. Glucose-6-phosphate dehydrogenase deficiency and
hematopoietic stem cell transplantation. Bone Marrow Transplant 29,
399-402 (2002). https://doi.org/10.1038/sj.bmt.1703369 PMid:11919729
- Pilo,
F. et al. Safety of hematopoietic stem cell donation in glucose 6
phosphate dehydrogenase-deficient donors. Bone Marrow Transplant 48,
36-39 (2013). https://doi.org/10.1038/bmt.2012.112 PMid:22732702