The erythroid-specific transcription factor KLF1 plays a critical role in HbF regulation by directly modulating BCL11A expression and recruiting chromatin-remodeling complexes to the β-globin locus.[5,9] Vinjamur et al. demonstrated that CRISPR-mediated disruption of KLF1 in primary human erythroblasts results in a 20–30% reactivation of γ-globin expression, accompanied by a reduction in BCL11A levels, a key mechanism underlying HbF silencing.[5] These findings highlight the therapeutic potential of targeting KLF1-dependent pathways, including pharmacological induction strategies such as hydroxyurea, as well as BCL11A-focused gene-editing approaches, underscoring the central role of KLF1 in HbF regulation and its relevance for SCA
management.[9-10]
Recent therapeutic strategies increasingly employ CRISPR-Cas9 technology to induce HbF re-expression. Elevated HbF levels have been shown to ameliorate disease severity in both β-thalassemia and SCA. BCL11A acts as a master repressor of γ-globin transcription, and disruption of its erythroid-specific enhancer within the second intron using CRISPR-Cas9 has been shown to markedly increase HbF levels, thereby reducing clinical manifestations of β-hemoglobinopathies.[11-13] CTX001 (exa-cel) is an ex vivo CRISPR-Cas9-based gene-editing therapy designed to reactivate HbF in autologous hematopoietic stem cells derived from patients with SCA or transfusion-dependent β-thalassemia. Ongoing clinical trials have shown encouraging safety and efficacy outcomes, supporting the potential of genome editing as a curative strategy for these disorders.[11-12]
Anemia, defined by reduced hemoglobin concentrations (<13.5 g/dL in men and <12.0 g/dL in women), affects more than 1.7 billion individuals worldwide and represents a major global health burden.[14-15] Anemia can have many causes; however, we can distinguish two fundamental forms: acquired and inherited.[14-15] Hemoglobinopathies are the most frequent monogenic diseases worldwide; it is estimated that 5% of the world’s population carries a defective hemoglobin (Hb) trait.[16-18] The most common hemoglobinopathies are β-thalassemia and sickle cell disease (SCD), both of which result from defects in the β-globin chain.[16-18] More than 40,000 infants are born with β-thalassemia each year, of whom about 25,500 have transfusion-dependent β-thalassemia, and an estimated 300,000 infants are born annually worldwide with Sickle Cell Disease (SCD).[16-18] Patients affected by β-thalassemia show low or absent production of adult β-globin chains, leading to α-globin/β-globin chain imbalance, erythroid cell death, hemolysis, and iron overload.[2,3] SCD is characterized by the production of a mutant β-globin chain (βS) that is incorporated in an Hb tetramer (HbS) that has a propensity to polymerize.[18] This polymerization causes red blood cell (RBC) sickling, hemolysis, vaso-occlusive crises (VOCs), and acute chest syndrome.[17,18] This review focuses on the genetic and epigenetic regulation of HbF in β-hemoglobinopathies, with particular attention to key regulatory polymorphisms at the BCL11A, XmnI, and HMIP-2 loci and their population-specific effects. In addition, emerging therapeutic strategies to increase HbF levels are discussed, with an emphasis on their implications for the management of β-hemoglobinopathies and related anemic conditions.
Biology of Hemoglobin (Hb) and Fetal Hemoglobin (HbF)
Two gene clusters on different chromosomes encode human hemoglobin: the β-globin cluster on chromosome 11 (which includes ε, Gγ, Aγ, δ, and β genes) and the α-globin cluster on chromosome 16 (which includes ζ and α genes).[19] These genes (Figure 1) are expressed in a specific sequence during development: embryonic hemoglobins (Hb Gower-1 [ζ₂ε₂], Hb Portland [ζ₂γ₂], and Hb Gower-2 [α₂ε₂]) are predominant early in gestation, fetal hemoglobin (HbF, α₂γ₂) becomes the primary hemoglobin after 8 weeks of gestation, and adult hemoglobins (HbA [α₂β₂] and HbA₂ [α₂δ₂]) dominate after birth.[20-21]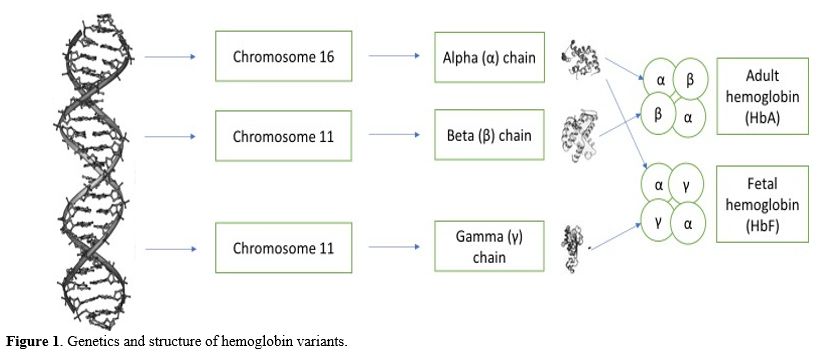 |
|
The γ-globin chains (Gγ and Aγ) differ by a single amino acid at position 136, glycine in Gγ versus alanine in Aγ.[22] At birth, HbF is composed of 70% Gγ and 30% Aγ, whereas in adults, this shifts to about 40% Gγ and 60% Aγ.[23] This change occurs during the γ-to-β globin switch, which is completed by 6-12 months of age.[23-24] HbF has a higher oxygen affinity (P50 = 19 mmHg) than HbA (P50 = 27 mmHg), thereby facilitating oxygen transfer from the mother to the fetus.[25-26] This difference comes from less binding of 2,3-BPG to γ-globin chains and altered hemoglobin-oxygen dissociation kinetics.[24]
In healthy adults, HbF is confined to F-cells, a subset of erythrocytes (1-5% of RBCs) that contain both HbF and HbA.[27] Unlike fetal RBCs, which exhibit macrocytic morphology with an MCV around 120 fL, adult F-cells maintain normal red blood cell size (MCV around 80 fL) and membrane properties.[27] During erythropoiesis, the HbF content per F-cell remains consistent, while HbA production gradually increases, demonstrating the precise regulation of globin genes during erythroid maturation.[22] The developmental γ-to-β globin switch represents the central biological process underlying HbF silencing and constitutes the primary therapeutic target in β-hemoglobinopathies.
Abnormal HbF persistence beyond infancy can result from various causes, including genetic conditions such as β-thalassemia, SCA, HPFH, KLF1, and mutations;[28-29] acquired disorders such as aplastic anemia, myelodysplastic syndromes, and erythropoietic stress;[30] and environmental exposures such as high altitude, smoking, and certain medications.[31] Modern laboratory techniques for HbF analysis include HPLC, which quantifies Hb fractions with over 95% accuracy, capillary electrophoresis to distinguish Gγ from Aγ chains,[32-33] flow cytometry to detect F-cells with 4-6 pg HbF per cell sensitivity, and mass spectrometry to identify rare Hb variants.[34-35]
Genetic Regulation of HbF
The genetic architecture controlling fetal hemoglobin production involves complex interactions among transcriptional regulators, chromatin modifiers, and locus control regions. Three primary genetic loci have been definitively identified as key modifiers of HbF levels through GWAS and functional genomic analyses.[5-7] The BCL11A gene on chromosome 2p16 encodes a zinc-finger transcription factor that serves as the primary regulator of γ-globin silencing.[36] Bauer and Orkin demonstrated that BCL11A functions as a molecular scaffold, mediating chromatin looping between the β-globin locus control region (LCR) and the adult β-globin promoter while physically displacing γ-globin genes from this active chromatin hub.[37] This mechanistic insight was derived from studies showing that BCL11A knockdown in adult erythroid cells reactivates γ-globin expression by 20-30%, particularly in cells carrying the rs11886868 (C→T) polymorphism in the BCL11A erythroid enhancer.[38,39] While BCL11A is the most extensively validated therapeutic target, the long-term consequences of its modulation, especially beyond the erythroid lineage, are not yet fully defined and may involve unintended effects on gene regulation, stem cell integrity, and broader physiological systems.The HBS1L-MYB intergenic region (HMIP-2) on chromosome 6q23 is the second central HbF regulatory locus.[40] Stadtholders et al. described this region as containing stage-specific enhancers that regulate MYB expression during erythropoiesis.[41] Through detailed haplotype analysis, Galarneau et al. identified rs9399137 (T→C) as the most significantly associated variant in European populations, accounting for 8-12% of HbF variability.[42] Functional studies in Tanzanian SCA patients showed that HMIP-2 variants influence the timing of MYB expression, thereby affecting the onset of erythroid differentiation and indirectly impacting γ-globin silencing.[43] In contrast to BCL11A, HMIP-2 effects are indirect and population-dependent, limiting their immediate therapeutic application and underscoring the need for further functional validation and mechanistic studies before they can be reliably translated into broadly effective clinical interventions.
The XmnI-HBG2 polymorphism (rs7482144) on chromosome 11p15 is the third primary HbF regulatory site. Cardoso et al. demonstrated that the C→T substitution at position -158 creates a new GATA-1 binding site,[44] which increases γ-globin transcription during stress erythropoiesis. This effect is particularly strong in carriers of the Arab-Indian β-globin haplotype, where the T allele frequency exceeds 80% and is associated with HbF levels of 30-40% in homozygous individuals.[45]
Epigenetic regulation adds another vital layer to HbF control. Xu et al. described how BCL11A recruits the NuRD chromatin remodeling complex to create repressive histone marks at the γ-globin promoters.[46] Another study that KLF1 directs this process by directly regulating BCL11A expression.[47] The potential of targeting these epigenetic mechanisms was demonstrated in clinical trials of histone deacetylase inhibitors, in which drugs such as sodium phenylbutyrate increased HbF by 15-20% by altering chromatin accessibility at the β-globin locus.[48] Another study[49] has identified the SPOP ubiquitin ligase as a new regulator of this epigenetic switch, providing new possibilities for targeted HbF induction. These polymorphisms account for 20-50% of the variance in HbF levels in β-thalassemia and SCA, making them targets for precision therapies.[27,36] Figure 2 illustrates the position of the 3 loci discussed in this section (Figure 3).
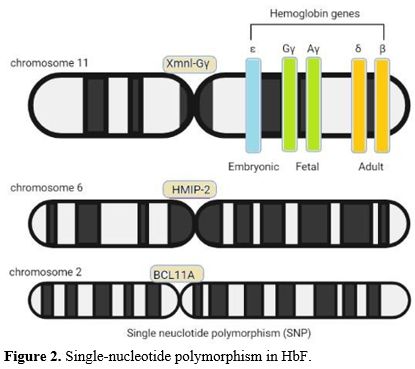 |
Figure 2. Single-nucleotide polymorphism in HbF. |
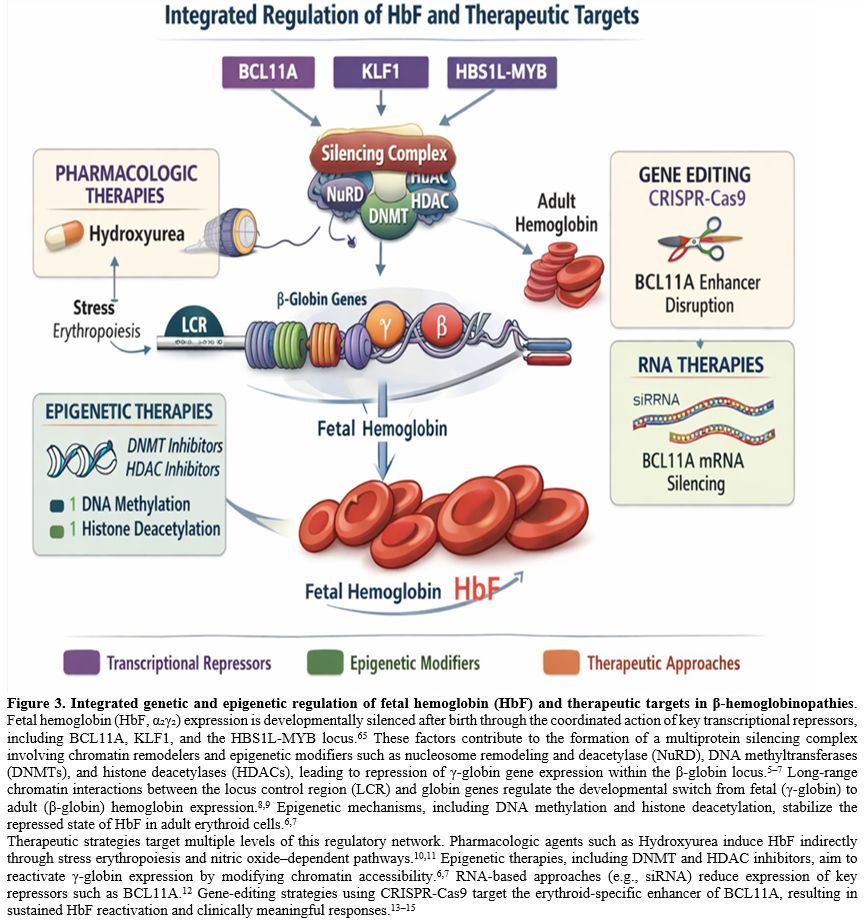 |
Figure 3. Integrated genetic and epigenetic regulation of fetal hemoglobin (HbF) and therapeutic targets in β-hemoglobinopathies.
Fetal hemoglobin (HbF, α₂γ₂) expression is developmentally silenced
after birth through the coordinated action of key transcriptional
repressors, including BCL11A, KLF1, and the HBS1L-MYB locus.[65] These
factors contribute to the formation of a multiprotein silencing complex
involving chromatin remodelers and epigenetic modifiers such as
nucleosome remodeling and deacetylase (NuRD), DNA methyltransferases
(DNMTs), and histone deacetylases (HDACs), leading to repression of
γ-globin gene expression within the β-globin locus.[5–7] Long-range
chromatin interactions between the locus control region (LCR) and
globin genes regulate the developmental switch from fetal (γ-globin) to
adult (β-globin) hemoglobin expression.[8,9] Epigenetic mechanisms,
including DNA methylation and histone deacetylation, stabilize the
repressed state of HbF in adult erythroid cells.[6,7] Therapeutic strategies target multiple levels of this regulatory network. Pharmacologic agents such as Hydroxyurea induce HbF indirectly through stress erythropoiesis and nitric oxide–dependent pathways.[10,11] Epigenetic therapies, including DNMT and HDAC inhibitors, aim to reactivate γ-globin expression by modifying chromatin accessibility.[6,7] RNA-based approaches (e.g., siRNA) reduce expression of key repressors such as BCL11A.[12] Gene-editing strategies using CRISPR-Cas9 target the erythroid-specific enhancer of BCL11A, resulting in sustained HbF reactivation and clinically meaningful responses.[13–15] |
Population-Specific Effects of HbF-Modifying Alleles
The regulation of fetal hemoglobin (HbF) varies significantly across different regions, influenced by differences in the distribution of genetic modifiers among populations (Table 2). Key alleles, such as the XmnI polymorphism (rs7482144) in the HBG2 promoter, are associated with higher HbF levels and reduced transfusion needs in patients with β-thalassemia.[50-51]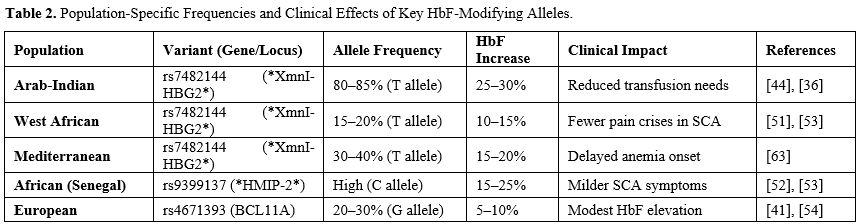 |
|
Genetic factors affecting HbF, such as the XmnI polymorphism, BCL11A variants (e.g., rs11886868), and HBS1L-MYB SNPs (like rs9399137), exhibit notable population-specific effects. Specific sickle cell hemoglobin haplotypes from Africa[52-54] and Southeast Asia, including the Senegal and Cameroon types, are associated with higher HbF levels, whereas European populations show distinct HMIP-2 patterns.[55] These ancestral variations, influenced by historical migration and selective pressures, suggest how genetic backgrounds shape HbF regulation and disease severity.[55] This underscores the need for developing population-specific therapies.[53]
BCL11A Polymorphism
The BCL11A gene on chromosome 2p16 acts as a key regulator of HbF silencing. It encodes a zinc-finger transcription factor that recruits the NuRD chromatin[56-57] remodeling complex to establish repressive histone marks at γ-globin promoters, effectively suppressing HbF expression in adult erythroid cells.[27,43] BCL11A serves as a molecular scaffold, facilitating long-range chromatin interactions between the β-globin locus control region (LCR) and the adult β-globin gene, while also displacing γ-globin genes from the active chromatin hub.[41,57] Significant SNPs like rs11886868 (C→T) in the BCL11A erythroid enhancer significantly diminish γ-globin suppression, with knockdown studies showing 20-30% reactivation of HbF.[58] This dose-dependent silencing activity makes BCL11A an appealing therapeutic target for β-hemoglobinopathies, as demonstrated by gene-editing techniques that selectively reduce BCL11A expression in erythroid cells, leading to increased HbF and reduced pathogenic HbS in sickle cell disease.The crucial roles of BCL11A in hematopoietic stem cell and B-lymphocyte development, as well as its overexpression in various hematological malignancies and solid tumors, have been documented, where it is associated with poor clinical outcomes.[57,59] This oncogenic activation may occur through two main mechanisms: genetic alterations, including viral integration, gene amplification, and chromosomal translocations, or epigenetic dysregulation involving microRNA suppression, abnormal long non-coding RNA activity, and transcription factor issues.[60-61] These findings position BCL11A not only as a key regulator of hemoglobin switching but also as a possible oncogenic driver in malignant transformation.[59]
HMIP-2 Polymorphism
The HBS1L-MYB intergenic region (HMIP-2) on chromosome 6q23 is another important HbF regulatory locus, though its mechanisms differ significantly from BCL11A.While the role of HBS1L is still unclear, the nearby MYB gene encodes c-MYB, a transcription factor that regulates erythroid proliferation and differentiation.[27,42,63] Variants in HMIP-2, such as rs9399137 (T→C) and rs4895441 (A→G), influence HbF levels by altering MYB expression during erythropoiesis, thereby indirectly affecting the γ-to-β globin switch.[63]
These SNPs account for 8-12% of HbF variability in European populations[51] and show distinct haplotype effects in African cohorts.[52] Unlike BCL11A direct repression, HMIP-2 variants influence HbF through stage-specific enhancers that extend erythroid progenitor proliferation, thereby delaying γ-globin silencing. This indirect regulation produces additive effects when co-occurring with BCL11A variants, as observed in Tanzanian patients with sickle cell disease, where specific HMIP-2 haplotypes are associated with milder disease.[63]
XmnI Polymorphism
The XmnI-HBG2 polymorphism (rs7482144, C→T) at position -158 of the γ-globin promoter is uniquely identified by its stress-responsive inducement of HbF. These variants forms a new GATA-1 binding site that enhances HBG2 transcription under erythropoietic stress, particularly in β-thalassemia and sickle cell anemia.[44] Its clinical effect varies significantly among populations whereby the Arab-Indian haplotypes[36] showed an 80-85% T allele frequency with a 25-30% increase in HbF and fewer transfusions needed,[64] whereas West African[53] and Mediterranean (30-40% frequency) groups show more moderate HbF rises of 10-15% and 15-20%, respectively.[64]The strong linkage of the polymorphism to the Arab-Indian β-globin haplotype explains its exceptional clinical benefits in these patients, who often maintain HbF levels above 30% when homozygous.[36] Unlike BCL11A and HMIP-2 variants, which operate through complex transcriptional networks, XmnI proximity to the promoter offers a more precise mechanism for HbF reactivation, making it a valuable marker for predicting disease severity.[64-65]
Therapeutic Strategies for HbF Induction
Identification of key regulators, including BCL11A, KLF1, and the HBS1L–MYB (HMIP-2) locus, has provided major mechanistic insights into developmental hemoglobin switching.[62] A seminal study has demonstrated that BCL11A functions as a master repressor of γ-globin expression, thereby establishing a molecular framework for therapeutic HbF reactivation.[66] In parallel, the identification of the XmnI-HBG2 polymorphism and mutations associated with hereditary persistence of fetal hemoglobin (HPFH) revealed naturally occurring genetic variants that sustain elevated HbF levels into adulthood, offering valuable biological models for targeted therapeutic strategies.[27,58,64]These discoveries directly facilitated the development of modern therapeutic approaches, including hydroxyurea, histone deacetylase (HDAC) and DNA methylation inhibitors, lentiviral gene addition, and, more recently, CRISPR–Cas9–mediated editing of the BCL11A erythroid enhancer and HPFH-mimicking promoter modifications.[67-68] The successful clinical translation of CRISPR-based therapies, particularly exa-cel, which reactivates HbF through targeted disruption of the BCL11A erythroid enhancer, clearly demonstrates that deciphering the genetic control of hemoglobin switching has enabled curative interventions for sickle cell disease and β-thalassemia.[60]
Pharmacological and genetic strategies to increase fetal hemoglobin (HbF) levels have been extensively investigated in patients with β-hemoglobinopathies. Cytotoxic agents such as hydroxyurea (HU) and 5-azacytidine interfere with DNA synthesis and stimulate HbF production, with HU demonstrating rapid clinical efficacy in a subset of patients.[52] This cytotoxic effect is associated with disruption of cell-cycle progression and induction of chromosomal instability in proliferating cells, as demonstrated in experimental models treated with vincristine and doxorubicin.[69] HDAC inhibitors modify chromatin architecture at the β-globin locus, resulting in approximately 15–20% increases in HbF.[70-71] More recently, selective inhibition of speckle-type POZ protein (SPOP) has emerged as a promising strategy, increasing HbF by preventing ubiquitin-dependent degradation of γ-globin activators and achieving efficacy comparable to HDAC inhibitors while avoiding genotoxicity.[72-73] However, while BCL11A remains the most validated therapeutic target, long-term safety of its modulation, particularly outside the erythroid lineage, remains incompletely defined.
Targeting BCL11A using precision genome-editing technologies offers exceptional potential for sustained HbF induction. CRISPR–Cas9 enables selective disruption of the erythroid-specific enhancer of BCL11A, leading to a marked reduction of BCL11A expression in erythroblasts while preserving its essential functions in non-erythroid lineages.[67-68] This strategy induces persistent HbF expression resembling HPFH, reduces globin chain imbalance, and mitigates hemolysis, making it a highly promising precision medicine approach for sickle cell disease and β-thalassemia.[27,52,64]
The clinical applicability of this approach is exemplified by exa-cel, an ex vivo CRISPR-edited autologous hematopoietic stem cell therapy developed for transfusion-dependent β-thalassemia (TDT) and severe sickle cell disease (SCD). Gene-edited CD34⁺ cells exhibit robust γ-globin induction, correction of globin chain imbalance, and sustained HbF elevation following reinfusion.[62,71,73] In pivotal trials and long-term follow-up studies reported by Frangoul et al., exa-cel consistently maintained HbF levels exceeding 40–45%, eliminated vaso-occlusive crises in SCD, and rendered the majority of β-thalassemia patients transfusion-independent.[11] These compelling outcomes led to regulatory approval of exa-cel in 2023–2024, marking the first approved CRISPR–Cas9–based therapy for β-hemoglobinopathies and a landmark achievement in genomic medicine.[67] Although exa-cel represents a major breakthrough, its applicability is currently restricted by cost, infrastructure, and long-term safety uncertainties.
Despite these advances, uncertainty remains regarding the phenotypic consequences of different KLF1 mutations in humans. Initial reports indicated that individuals carrying specific missense mutations exhibited elevated HbF levels ranging from 3% to 19% of total hemoglobin.[74] However, subsequent studies of heterozygous KLF1 mutations revealed either disrupted erythropoiesis or minimal effects on HbF expression.[75] Elucidating the basis of this variability is essential for understanding the direct and indirect roles of KLF1 in HbF regulation and for evaluating its feasibility as a therapeutic target without compromising erythroid differentiation.
Similarly, SOX6 has emerged as a potential HbF regulator; however, its essential role in erythropoiesis complicates therapeutic targeting. Notably, heterozygous disruption of SOX6 in humans failed to induce HbF, suggesting the existence of dosage compensation mechanisms or a requirement for more profound suppression to achieve meaningful HbF induction.[76-77] Beyond gene editing, thalidomide and its derivatives have attracted interest as pharmacological HbF inducers. In vitro studies of ineffective erythropoiesis indicate that thalidomide can enhance γ-globin mRNA expression in a dose-dependent manner by modulating transcription factors such as BCL11A, SOX6, GATA1, and KLF1, as well as through p38 MAPK–mediated post-translational mechanisms.[78-80]
Pomalidomide, a third-generation immunomodulatory derivative with a more favorable safety profile, has demonstrated robust HbF induction in models of β-thalassemia, HbE disease, and sickle cell anemia. Comparable to hydroxyurea, pomalidomide increased HbF levels without inducing myelosuppression in humanized SCD mouse models.[81] Furthermore, treatment of hematopoietic stem cells with pomalidomide or lenalidomide significantly enhanced stem cell proliferation and HbF induction via transcriptional regulation of HBB and HBG, accompanied by downregulation of repressors including BCL11A, IKZF1, KLF1, LSD1, and SOX6.[79-80]
Additional mechanistic insights were provided by Lechauve et al., who demonstrated that the autophagy-activating kinase ULK1 plays a pivotal role in clearing excess free α-globin chains. In β-thalassemic mouse models, loss of ULK1 impaired autophagy, exacerbated disease severity, and hindered α-globin clearance, whereas pharmacological activation of ULK1 enhanced autophagy and reduced toxic α-globin accumulation in erythroid precursors.[82]
Finally, Mettananda et al. employed CRISPR–Cas9 to downregulate α-globin expression by deleting the MCS-R2 α-globin enhancer, mimicking a naturally occurring α-thalassemia mutation.[83] This approach corrected globin chain imbalance in gene-edited CD34⁺ cells derived from β-thalassemia patients.[67] Subsequent work by Pavani et al. confirmed that targeted editing of the α-globin locus, including deletion of HBA2, induces a mild α-thalassemia trait that restores α/β-globin balance and effectively rescues the β-thalassemia phenotype.[84]
Overall, HbF induction strategies can be stratified into (i) clinically validated approaches (hydroxyurea, exa-cel), (ii) advanced clinical development (epigenetic modifiers), and (iii) emerging experimental platforms (base/prime editing, epigenome engineering). This distinction is critical for interpreting current translational relevance. (Table 1, Figure 3)
Clinical Implications and Future Directions
Modulating fetal hemoglobin (HbF) represents a transformative strategy for the treatment of β-hemoglobinopathies, with advances in human genetics increasingly enabling personalized therapeutic approaches. Population-specific polymorphisms, including XmnI, BCL11A, and HBS1L–MYB (HMIP-2), underscore the necessity for tailored interventions, as their influence on HbF levels varies substantially across ethnic groups.[5-7] For instance, clinical responsiveness to hydroxyurea differs markedly among patients with distinct genetic backgrounds, highlighting the importance of pharmacogenomic profiling in optimizing treatment outcomes.[52] Future investigations should prioritize rational combination strategies such as integrating HDAC inhibitors with genome-editing approaches to maximize HbF induction while minimizing toxicity.[70]In parallel, the development of non-invasive biomarkers capable of monitoring HbF dynamics and enabling early intervention in high-risk populations, including pregnant women with anemia, may further enhance clinical outcomes.[85-86] By integrating genetic, epigenetic, and clinical datasets, next-generation therapeutic frameworks may achieve sustained HbF elevation and ultimately reduce the global burden of hemoglobinopathies.[87]
Emerging next-generation CRISPR technologies, including base editing and prime editing, offer highly precise methods for HbF reactivation without generating double-strand DNA breaks. Cytosine and adenine base editors enable single-nucleotide substitutions that recapitulate naturally occurring HPFH-associated mutations within the HBG1/2 promoters, resulting in persistent γ-globin expression with minimal genomic injury.[58,73] Prime editing further extends this capability by enabling programmable insertions, deletions, or nucleotide substitutions within γ-globin promoters and regulatory elements, thereby allowing accurate reconstruction of HPFH-like variants and selective disruption of repressor-binding motifs. Early preclinical studies indicate that these precision-editing approaches can induce robust HbF expression while preserving hematopoietic stem cell integrity, positioning next-generation CRISPR systems as highly promising therapeutic platforms for β-hemoglobinopathies.[88] However, these approaches remain largely preclinical, and their clinical relevance is still to be established.
Beyond single-gene targeting, multiplex genome editing enables the simultaneous modification of multiple regulatory elements governing HbF expression. Coordinated editing of the BCL11A erythroid enhancer, HBG promoters, and key erythroid transcription factor binding sites have been shown to produce synergistic increases in γ-globin levels.[88-89] Such combinatorial strategies more closely emulate the complex regulatory architecture underlying hemoglobin switching and may better accommodate patient- or mutation-specific variab
ility in HbF responsiveness. In addition to genome editing, epigenome engineering approaches employing dCas9-based systems allow modulation of chromatin states at fetal globin loci without altering the underlying DNA sequence. For example, dCas9–KRAB or dCas9–DNMT3A can repress HbF silencers such as BCL11A or ZBTB7A, whereas dCas9–p300 and dCas9–TET1 can remodel chromatin to enhance HBG transcription.[90-91] These reversible, programmable strategies offer potentially safer alternatives to permanent genome modification and further expand the therapeutic landscape for HbF reactivation in β-hemoglobinopathies.
Conclusions
The regulation of HbF is a key therapeutic target in managing β-hemoglobinopathies, including SCA and β-thalassemia. This review emphasizes the critical roles of genetic and epigenetic mechanisms, particularly polymorphisms in BCL11A, HMIP-2, and XmnI-HBG2, in influencing HbF levels across diverse populations. These variants explain 20–50% of HbF variability and are associated with clinical outcomes, including decreased transfusion requirements in Arab and Indian β-thalassemia carriers and milder SCA symptoms in African populations. Pharmacological agents such as hydroxyurea and HDAC inhibitors have been shown to increase HbF levels, although individual responses vary due to genetic differences. New gene-editing techniques, such as BCL11A knockdown, offer promising options for targeted therapy by reactivating γ-globin expression while reducing off-target effects. Nonetheless, challenges persist, including the need for lineage-specific targeting and the development of optimized combination treatments to improve both efficacy and safety.Abbreviations
Hb: Hemoglobin, HbF: Fetal hemoglobin, HbA: Adult hemoglobin, SCA: Sickle cell anemia, HPFH: Hereditary persistence of fetal hemoglobin, HU: Hydroxyurea, SNPs: Single-nucleotide polymorphisms, HDAC: Histone deacetylase, GWAS: Genome-wide association study.Authors’ Contribution
Conceptualization: Yousef Saeed Mohammad Abu Za’ror; Methodology: Yousef Saeed Mohammad Abu Za’ror, Fatima Azzahra Delmani, Maryam Azlan, Jehad Farouq Alhmoud, Amer Mohammad Ayasreh, Sarah Ihsan Al-wendawi; Formal analysis and investigation: Yousef Saeed Mohammad Abu Za’ror, Maryam Azlan; Writing - original draft preparation: Yousef Saeed Mohammad Abu Za’ror, Fatima Azzahra Delmani; Writing - review and editing: Yousef Saeed Mohammad Abu Za’ror, Fatima Azzahra Delmani, Maryam Azlan, Joseph Bagi Suleiman, Jehad Farouq Alhmoud, Amer Mohammad Ayasreh, Sarah Ihsan Al-wendawi; Resources: Yousef Saeed Mohammad; Supervision: Maryam Azlan.Data Availability Statement
All are available upon reasonable request.References
- Ahmed, MH, Ghatge, MS, & Safo, MK. (2020). Hemoglobin:
Structure, Function and Allostery. Subcellular Biochemistry, 94,
345–382. https://doi.org/10.1007/978-3-030-41769-7_14
- Marengo-Rowe AJ. Structure-function relations of human hemoglobins. Proc (Bayl Univ Med Cent). 2006;19(3):239-245. https://doi.org/10.1080/08998280.2006.11928171
- Wang X, Thein SL. Switching from fetal to adult hemoglobin. Nat Genet. 2018;50(4):478-480. https://doi.org/10.1038/s41588-018-0094-z
- Sethi M, Goyal L, Gupta S, Kumar R. Markedly Elevated Fetal
Hemoglobin in Myeloid Neoplasms: A Potential Diagnostic Pitfall during
Hemoglobinopathy Work-Up. J Lab Physicians. 2025; [Epub ahead of
print]. https://doi.org/10.25259/JLP_313_2025
- Vinjamur DS, Bauersachs HG, Bauer DE. Genetic and epigenetic modifiers of fetal hemoglobin. Front Genet. 2021;12:656143. https://doi.org/10.3389/fgene.2021.656143
- Thein SL. Genetic modifiers of fetal hemoglobin levels in health
and disease. Cold Spring Harb Perspect Med. 2013;3(12):a011700. https://doi/org/10.1101/cshperspect.a011700
-
Menzel S, Garner C, Gut I, Sappert C, Rosen-Wolff A, Broxholme J, et
al. A QTL influencing F cell production maps to a gene-rich locus on
chromosome 2p16.1. Nat Genet. 2007;39(10):1197-1199. https://doi.org/10.1038/ng2108
-
Al-Kindi S, Al-Zadjali S, Al-Haddabi H, Al-Riyami N, Al-Mushaikhi Z,
Pathare A. Single Nucleotide Polymorphisms in XMN1-HBG2, HBS1L-MYB, and
BCL11A and Their Relation to High Fetal Hemoglobin Levels That
Alleviate Anemia. Diagnostics (Basel). 2022;12(6):1374. https://doi.org/10.3390/diagnostics12061374
-
Caria CA, Faa V, Ristaldi MS. Kruppel-Like Factor 1: A Pivotal Gene Regulator in Erythropoiesis. Cells. 2022;11(19):3097. https://doi.org/10.3390/cells11193097
-
Kanter J, Kruse-Jarres R, Bernaudin F. Sickle cell disease. Lancet.
2023;401(10381):1025-1039. https://doi.org/10.1016/S0140-6736(22)02381-8
-
Frangoul H, Locatelli F, Sharma A, Bhatia M, Mapara M, Molinari L, et
al. Exagamglogene Autotemcel for Severe Sickle Cell Disease. N Engl J
Med. 2024;390(18):1649-1662. https://doi.org/10.1056/NEJMoa2309675
-
Locatelli F, Lang P, Wall D, Meisel R, Corbacioglu S, Li AM, et al.
Exagamglogene Autotemcel for Transfusion-Dependent β-Thalassemia. N
Engl J Med. 2024;390(18):1663-1676.
https://doi.org/10.1056/NEJMoa2309673
-
Huang P, Wang Y, Xu C, et al. Silencing of BCL11A by disrupting
enhancer-dependent epigenetic insulation. Blood.
2026;147(13):1470-1485. https://doi.org/10.1182/blood.2025028411
-
GBD 2021 Anaemia Collaborators. Prevalence, causes, and consequences of
anaemia: a systematic analysis for the Global Burden of Disease Study
2021. Lancet Haematol. 2023;10(9):e713-e734.
https://doi.org/10.1016/S2352-3026(23)00160-4
-
Safiri S, Kolahi AA, Noori M, Nejadghaderi SA, Karamzad N, Bragazzi NL,
et al. Burden of anemia and its underlying causes in 204 countries and
territories, 1990-2019: results from the Global Burden of Disease Study
2019. J Hematol Oncol. 2021;14(1):185.
https://doi.org/10.1186/s13045-021-01202-2.
-
De Sanctis V, Kattamis C, Canatan D, Soliman AT, Elsedfy H, Karimi M,
Daar S, Wali Y, Yassin M, Soliman N, Sobti P, Al Jaouni S, El Kholy M,
Fiscina B, Angastiniotis M. β-Thalassemia Distribution in the Old
World: an Ancient Disease Seen from a Historical Standpoint. Mediterr J
Hematol Infect Dis. 2017 ;9(1):e2017018.
https://doi.org/10.4084/MJHID.2017.018
-
Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A Review. JAMA. 2022; 328(1):57-68. https://doi.org/10.1001/jama.2022.10233
-
Magrin E, Miccio A, Cavazzana M. Lentiviral and genome-editing
strategies for the treatment of β-hemoglobinopathies. Blood. 2019;
134(15):1203-1213. https://doi.org/10.1182/blood.2019000949
-
Tesio N, Bauer DE. Molecular basis and genetic modifiers of
thalassemia. Hematol Oncol Clin North Am. 2023;37(2):273-299.
https://doi.org/10.1016/j.hoc.2022.12.001
-
Ahmed MH, Ghatge MS, Safo MK. Hemoglobin: Structure, Function and
Allostery. Subcell Biochem. 2020;94:345-382.
https://doi.org/10.1007/978-3-030-41769-7_14
-
Pellegrino C, Stone EF, Valentini CG, Teofili L. Fetal Red Blood Cells:
A Comprehensive Review of Biological Properties and Implications for
Neonatal Transfusion. Cells. 2024 Nov 7;13(22):1843.
https://doi.org/10.3390/cells13221843
-
Hardison RC. Mechanisms of Globin Gene Regulation in Mammals. Annu Rev
Genet. 2025 Aug
19;59:341-367. https://doi.org/10.1146/annurev-genet-020325-095743
-
Bagchi A, Billakanti S, Shehu V, Khandros E. Switching and Sniffing
around the β-globin cluster. Blood. 2026 Feb 5;147(6):609-610.
https://doi.org/10.1182/blood.2025028411
-
Ayuba S, Bashar F, Abbas A. Targeting fetal hemoglobin induction in
sickle cell anemia: Epigenetic and gene-modifying therapeutic
strategies. Int J Epigenetics. 2025 Nov;5(1):1-6.
https://doi.org/10.3892/ije.2025.29
-
Blain L, Watier C, Weng X, Masse A, Bédard MJ, Bettache N, et al.
Prospective Evaluation of Fetal Hemoglobin Expression in Maternal
Erythrocytes: An Analysis of a Cohort of 345 Parturients. Diagnostics
(Basel). 2023 May 27;13(11):1873.
https://doi.org.10.3390/diagnostics13111873
-
Hussein KS. The role of fetal hemoglobin in predicting preeclampsia in
early pregnancy. Life Sci J. 2018;15(1):60-66.
https://doi.org/10.7537/marslsj150118.08
-
Sankaran VG, Weiss MJ. Fetal hemoglobin in health and disease. Blood.
2024 Jan 11;143(2):109-120. https://doi.org/10.1182/blood.2023020684
-
Wang, X. and S.L. Thein, Switching from fetal to adult hemoglobin.
Nature genetics, 2018. 50(4): p. 478.
https://doi.org/10.1038/s41588-018-0094-z
-
Bhanushali, A.A., et al., Genetic variant in the BCL11A (rs1427407),
but not HBS1-MYB (rs6934903) loci associate with fetal hemoglobin
levels in Indian sickle cell disease patients. Blood Cells, Molecules,
and Diseases, 2015. 54(1): p. 4-8.
https://doi.org/10.1016/j.bcmd.2014.10.003
-
Lolis, D., et al., High HbF in pregnancy is associated with the Xmn I
polymorphism at the− 158bp of the Gγ-globin gene. European Journal of
Obstetrics & Gynecology and Reproductive Biology, 1995. 60(2): p.
153-156. https://doi.org/10.1016/0028-2243(95)02105-2
-
Sankaran VG, Power C, Xu J, Brunner MC, Lettre G, Stephens AS, et al.
Developmental globin switching driven by BCL11A. Nature. 2009 Oct
29;460(7259):1093-1097. https://doi.org/10.1038/nature08263
-
Greene DN, Junker ET, Toffaletti JG. Interpreting Hemoglobin
Electrophoresis and HPLC. Clin Chem. 2023 Nov 2;69(11):1224-1234.
https://doi.org/10.1093/clinchem/hvad131
-
Shook LM, Haygood D, Quinn CT. Laboratory Diagnosis of
Hemoglobinopathies. Cold Spring Harb Perspect Med. 2021 Aug
2;11(8):a039032. https://doi.org/10.1101/cshperspect.a039032
-
Eliasen R, Shah A, Smith A, et al. Comparison of Sickle Solubility Test
with Mass Spectrometry for Hemoglobin S Confirmation. Hemoglobin.
2025;49(1):12-18. https://doi.org/10.1080/03630269.2025.2595002
-
Bellad A, Rangiah K, Chavan S, Warade J, Das B, Pandey A. A Mass
Spectrometry–Based Multiplexed Targeted Assay for Detection of
Hemoglobinopathies from Dried Blood Spots. J Mol Diagn. 2025
Jan;27(1):45-56. https://doi/org.10.1016/j.jmoldx.2024.10.007
-
Vathipadiekal V, Farrell JJ, Wang S, Jia L, Pang H, Carranza GC, et al.
Homozygosity for a haplotype in the HBG2-OR51B4 region is exclusive to
Arab-Indian haplotype sickle cell anemia. Am J Hematol. 2011
Apr;86(4):356-359. https://doi.org/10.1002/ajh.21976
-
Bauer DE, Orkin SH. Hemoglobin switching's surprise: the versatile
BCL11A gene. Curr Opin Genet Dev. 2015;33:62–70.
https://doi.org/10.1016/j.gde.2015.08.003
-
Bae HT, Baldwin CT, Sebastiani P, Milton JN, Hartley SW, Beiswanger C,
et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and
HBS1L-MYB are major modifiers of HbF in African Americans. Am J
Hematol. 2012 Apr;87(4):396-399. https://doi.org/10.1002/ajh.23125
-
Bhanushali AA, Das BR, Verma IC. Genetics of fetal hemoglobin in tribal
Indian patients with sickle cell anemia. Transl Res. 2015
Dec;166(6):783-787. https://doi/org.10.1016/j.trsl.2015.01.002
-
Wahlberg K, Jiang J, Rooks H, Howard J, Best S, Spector TD, et al. The
HBS1L-MYB intergenic interval associated with fetal hemoglobin levels
and age-related decline in HbF contains complex regulatory elements.
Blood. 2009 Aug 6;114(6):1254-1262.
https://doi.org/10.1182/blood-2009-03-210138
-
Stadhouders R, Aktas N, Thongjuea S, Aghajanirefah A, Siegers JY,
Farwick M, et al. The HBS1L-MYB intergenic locus regulates erythrocyte
traits, hemoglobin synthesis and sickle cell disease severity by
controlling MYB expression. Nat Commun. 2014 May 29;5:3948.
https://doi.org/10.1038/ncomms4948
-
Galarneau G, Palmer CD, Sankaran VG, Lettre G, Howard J, Hadley JS, et
al. The HLA-HFE region is a modifier of fetal hemoglobin levels in
sickle cell disease. Nat Genet. 2010 Dec;42(12):1049-1051.
https://doi.org/10.1038/ng.713
-
Xu J, Peng C, Sankaran VG, Shao Z, Esrick EB, Fedistiova BG, et al.
Corepressor-dependent silencing of fetal hemoglobin expression by
BCL11A. Proc Natl Acad Sci U S A. 2013 Apr 16;110(16):6518-6523.
https://doi.org/10.1073/pnas.1303976110
-
Cardoso WA, de Azevedo SCL, de Araujo-Sousa RF, Ferreira MB, da
Costa-Gomes AB, Junior CLC, et al. GATA-1 and GATA-2 transcription
factors in the regulation of the human β globin locus. Sci Rep. 2017
Feb 22;7:43210. https://doi.org/10.1038/srep43210
-
Barbosa IL, Silva MC, Santos MN, Goncalves MS, Araujo-Sousa RF,
Ferreira MB, et al. European chromosome 6 haplotypes augment fetal
hemoglobin levels in Brazilian sickle cell anemia patients. Blood. 2012
Oct 4;120(14):2917-2918. https://doi.org/10.1182/blood-2012-07-440404
-
Xu J, Bauer DE, Kerenyi MA, Vo TD, Hou S, Hsu YJ, et al.
Corepressor-dependent silencing of fetal hemoglobin expression by
BCL11A. Proc Natl Acad Sci U S A. 2013 Apr 16;110(16):6518-6523.
https://doi.org/10.1073/pnas.1303976110.
-
Paikari A, Sheehan VA. Fetal haemoglobin induction in sickle cell
disease. Br J Haematol. 2018 Jan;180(2):189-200.
https://doi.org/10.1111/bjh.14993
-
Zakaria NA, Islam MA, Abdullah WZ, Bahar R, Yusoff AAM, Wahab RA, et
al. Epigenetic Insights and Potential Modifiers as Therapeutic Targets
in -Thalassemia. Biomolecules. 2021 May 18;11(5):755.
https://doi.org/10.3390/biom11050755
-
Lee JS, Lee JK, Shin HJ, Shin E, Xu J, Bauer DE, et al. The E3 ligase
adaptor molecule SPOP regulates fetal hemoglobin levels in adult
erythroid cells. Blood. 2019 Sep 19;134(12):961-973.
https://doi.org/10.1182/blood.2019001153
-
Leonardo FC, Menzel S, Brugnerotto AF, Fertrin KY, Bezerra MAC, Araujo
AS, et al. European chromosome 6 haplotypes significantly augment fetal
hemoglobin levels in Brazilian sickle cell anemia patients: influence
of four HBS1L-MYB intergenic region SNPs. Blood. 2012 Nov
15;120(21):1002. https://doi.org/10.1182/blood.V120.21.1002.1002
-
Vadolas J, Nualkaew T, Voon HPJ, Vilcassim S, Grigoriadis G. Interplay
between α-thalassemia and β-hemoglobinopathies: Translating
genotype–phenotype relationships into therapies. Hemasphere. 2024 May
15;8(5):e78. https://doi.org/10.1002/hem3.78
-
Amuzu EX, Urio F, Dogbe EE, Ponsian P, Abubakar SY, Okeke C, et al.
Clinical manifestations of sickle cell disease in Africa and its
association with foetal haemoglobin parameters. Commun Med. 2025 Jun
18;5:238. https://doi.org/10.1038/s43856-025-00954-z
-
Nkya S, Makani J, Flanagan JM. Genetics and genomics in sickle cell
disease in Africa. Am J Hematol. 2026 Mar 14;101(Suppl 1):47-55.
https://doi.org/10.1002/ajh.70220
-
Wonkam A, Esoh K, Levine RM, Ngo Bitoungui VJ, Mnika K, Nimmagadda N,
et al. FLT1 and other candidate fetal haemoglobin modifying loci in
sickle cell disease in African ancestries. Nat Commun. 2025 Mar
1;16:2092. https://doi.org/10.1038/s41467-025-56432-1
-
Lee JS, Cho SI, Park SS, Seong MW. Molecular basis and diagnosis of
thalassemia. Blood Res. 2021 Apr 30;56(S1):S39-S43.
https://doi.org/10.1045/br.2021.2020332
-
Sher F, Hossain M, Seruggia D, Schoonenberg VAC, Yao Q, Cifani P, et
al. Rational targeting of a NuRD subcomplex guided by comprehensive in
situ mutagenesis. Nat Genet. 2019 Jul;51(7):1149-1159.
https://doi.org/10.1038/s41588-019-0453-4
-
Zheng G, Yin M, Mehta S, Chu IT, Wang S, AlShaye A, et al. A tetramer
of BCL11A is required for stable protein production and fetal
hemoglobin silencing. Science. 2024 Nov 28;386(6725):1010-1018.
https://doi.rg/10.1126/science.adp3025
-
Viennet T, Yin M, Jayaraj A, Kim W, Sun ZYJ, Fujiwara Y, et al.
Structural insights into the DNA-binding mechanism of BCL11A: the
integral role of ZnF6. Structure. 2024 Oct 17;32(12):2276-2286.e4.
https://doi.org/10.1016/j.str.2024.09.022
-
Zhang H, Zeng J, Zhang F, Liu J, Liang L. Role of B-cell
lymphoma/leukemia 11A in normal and malignant hematopoiesis. Biology.
2025 Jan 1;14(1):26. https://doi.org/10.3390/biology14010026
-
Wang H, Chen M, Xu S, Pan Y, Zhang Y, Huang H, et al. Abnormal
regulation of microRNAs and related genes in pediatric β-thalassemia. J
Clin Lab Anal. 2021 Aug 16;35(9):e23945.
https://doi.org/10.1002/jcla.23945
-
Lulli V, Romania P, Morsilli O, Cianciulli P, Gabbianelli M, Testa U,
et al. MicroRNA-486-3p regulates γ-globin expression in human erythroid
cells by directly modulating BCL11A. PLoS One. 2013 Apr 4;8(4):e60436.
https://doi.org/10.1371/journal.pone.0060436
-
Nkya S, Makani J, Flanagan JM. Genetics and genomics in sickle cell
disease in Africa. Am J Hematol. 2026 Mar 14;101(Suppl 1):47-55.
https://doi.org/10.1002/ajh.70220
-
Mtatiro G, Mgaya J, Singh T, Menzel S, Cox SE, Soka D, et al. Genetic
association of fetal-hemoglobin levels in individuals with sickle cell
disease in Tanzania maps to conserved regulatory elements within the
MYB core enhancer. BMC Med Genet. 2015 May 5;16:31.
https://doi.org/10.1186/s12881-015-0174-8
-
Thein SL. Genetic modifiers of the β-haemoglobinopathies. Br J
Haematol. 2008 May;141(3):357-366.
https://doi.org/10.1111/j.1365-2141.2008.07025.x
-
Galanello R, Origa R. β-thalassemia. Orphanet J Rare Dis. 2010 May 21;5:11. https://doi.org/10.1186/1750-1172-5-11
-
Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Baena E, et al. Human
fetal hemoglobin expression is regulated by the zinc finger-protein
BCL11A. Science. 2008 Dec 19;322(5909):1839-1842.
https://doi.org/10.1126/science.1165409
-
Frangoul H, Altshuler D, Cappellini MD, Chen YS, Domm J, Eustace BK, et
al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia.
N Engl J Med. 2021 Jan 21;384(3):252-260.
https://doi.org/10.1056/NEJMoa2031054
-
Tesio N, Bauer D. Molecular basis and genetic modifiers of thalassemia.
Hematol Oncol Clin North Am. 2023 Apr;37(2):273-299.
https://doi.org/10.1016/j.hoc.2022.12.001
-
Almuhur R, Aljamal A, Al Shawabkeh M, Delmani FA, Alqadi T, Khwaldeh A.
A comparative study of mitotic index and chromosome aberrations in
vincristine and doxorubicin-treated normal female mice. Int J
Pharmacol. 2025;21(4). https://doi.org/10.3923/ijp.2024.115.120
-
Jha BK, Saunthararajah Y. Epigenetic modifier directed therapeutics to
unleash healthy genes in unhealthy cells. Semin Hematol. 2021
Jan;58(1):1-3. https://doi.org/10.1053/j.seminhematol.2021.01.001
-
Bou-Fakhredin R, De Franceschi L, Motta I, Cappellini MD, Taher AT.
Pharmacological induction of fetal hemoglobin in β-thalassemia and
sickle cell disease: an updated perspective. Pharmaceuticals (Basel).
2022 Jun 16;15(6):753. https://doi.org/10.3390/ph15060753
-
Lan X, Khandros E, Huang P, Peslak SA, Bhardwaj SK, Grevet JD, et al.
The E3 ligase adaptor molecule SPOP regulates fetal hemoglobin levels
in adult erythroid cells. Blood Adv. 2019 May 28;3(10):1586-1597.
https://doi.org/10.1182/bloodadvances.2019032318
-
Khandros E, Blobel GA. Elevating fetal hemoglobin: recently discovered
regulators and mechanisms. Blood. 2024 May 12;144(8):845-852.
https://doi.org/10.1182/blood.2023022190
-
Arnaud L, Seasonal C, Brunk BP, et al. A dominant mutation in the gene
encoding the erythroid transcription factor KLF1 causes a congenital
dyserythropoietic anemia. Am J Hum Genet. 2010;87(5):721-727.
https://doi.org/10.1016/j.ajhg.2010.10.010
-
Caria CA, Faà V, Ristaldi MS. Krüppel-like factor 1: a pivotal gene
regulator in erythropoiesis. Cells. 2022 Sep 29;11(19):3069.
https://doi.org/10.3390/cells11193069
-
Yi Z, Cohen-Barak O, Hagiwara N, Kingsley PD, Fuchs DA, Erickson DT, et
al. Sox6 directly silences epsilon globin expression in definitive
erythropoiesis. PLoS Genet. 2006 Feb 3;2(2):e14.
https://doi.org/10.1371/journal.pgen.0020014
-
Li J, Lai Y, Luo J, Luo L, Liu R, Liu Z, et al. SOX6 downregulation
induces γ-globin in human β-thalassemia major erythroid cells. Biomed
Res Int. 2017 Nov 28;2017:9496058. https://doi.org/10.1155/2017/9496058
-
Li X, Hong W, Wu J, Liu Q, Liu Y, Hu S, et al. Clinical patterns of
thalidomide in the treatment of transfusion-dependent β-thalassaemia in
children: a prospective single-arm study in China. Ann Med.
2025;57(1):2561219. https://doi.org/10.1080/07853890.2025.2561219
-
Yasara N, Thilakarathne S, Perera KDC, Premawardhena A, Mettananda S.
Efficacy and safety of thalidomide in $\beta$-thalassaemia: a
systematic review and meta-analysis. Sci Rep. 2026 Apr 3.
https://doi.org/10.1038/s41598-026-46504-y
-
Aradhya A, Javaid A, Oganesyan A, Fidler C. Thalidomide in
beta-thalassemia – a systematic review and meta-analysis. Blood. 2025
Nov;146(Suppl 1):6477. https://doi.org/10.1182/blood-2025-6477
-
Meiler SE, Wade M, Kutlar F, et al. Pomalidomide augments fetal
hemoglobin production without the myelosuppression of hydroxyurea in
mice with sickle cell disease. Blood. 2011;118(4):1109-1112.
https://doi.org/10.1182/blood-2011-03-340265.
-
Lechauve C, Keith J, Khandros E, Fowler S, Mayberry K, Low PS, et al.
The autophagy-activating kinase ULK1 mediates clearance of free
α-globin in β-thalassemia. Sci Transl Med. 2019 Mar
13;11(483):eaav4881. https://doi.org/10.1126/scitranslmed.aav4881
-
Mettananda S, et al. Editing an -globin enhancer in primary human
hematopoietic stem cells to treat $\beta$-thalassaemia. Nat Commun.
2017;8:15451. https://doi.org/10.1038/ncomms15451
-
Pavani G, Fabiano A, Laurent M, Amor F, Cantú C, Chalumeau A, et al.
Correcting β-thalassemia by CRISPR/Cas9 editing of the α-globin locus
in human hematopoietic stem cells. Nat Commun. 2021 Jun 17;12(1):3738.
https://doi.org/10.1038/s41467-021-23963-4
-
Ratriana T, Ginanjar E, Widajanti N, Gatot D, Setianingsih I, Sjakti
HA, et al. Modifying effects of XmnI, BCL11A, and HBS1L-MYB on clinical
appearance in β-thalassemia and HbE/β-thalassemia patients in
Indonesia. Hematol Oncol Stem Cell Ther. 2016 Dec;9(4):147-154.
https://doi.org/10.1016/j.hemonc.2016.08.001
-
Lai Y, Zhou L, Yi S, Chen Y, Tang Y, Yi S, et al. The association
between four SNPs (rs7482144, rs4671393, rs28384513 and rs4895441) and
fetal hemoglobin levels in Chinese Zhuang β-thalassemia intermedia
patients. Blood Cells Mol Dis. 2017 Mar;63:52-57.
https://doi.org/10.1016/j.bcmd.2017.01.011
-
Sharma A, Boelens JJ, Cancio M, Hankins JS, Bhad P, Azizy M, et al.
CRISPR-Cas9 Editing of the HBG1 and HBG2 Promoters to Treat Sickle Cell
Disease. N Engl J Med. 2023 Aug 30;389(9):820-832.
https://doi.org/10.1056/NEJMoa2215643
-
Liu N, Xu S, Yao Q, Zhu Q, Kai Y, Hsu JY, et al. Transcription factor
competition at the γ-globin promoters controls hemoglobin switching.
Nat Genet. 2021 Mar;53(4):511-520.
https://doi.org/10.1038/s41588-021-00798-y
-
Han W, Qiu HY, Sun S, Fu ZC, Wang GQ, Qian X, et al. Base editing of
the HBG promoter induces potent fetal hemoglobin expression with no
detectable off-target mutations in human HSCs. Cell Stem Cell. 2023 Dec
7;30(12):1624-1639.e8. https://doi.org/10.1016/j.stem.2023.10.007
-
Fontana L, Alahouzou Z, Miccio A, Antoniou P. Epigenetic regulation of
β-globin genes and the potential to treat hemoglobinopathies through
epigenome editing. Genes (Basel). 2023 Feb 25;14(3):577.
https://doi.oorg/10.3390/genes14030577
- Amistadi S, Fontana L, Magnoni C, Felix T, Charvin MK, Martinucci P, et al. Dissecting the epigenetic regulation of the fetal hemoglobin genes to unravel a novel therapeutic approach for β-hemoglobinopathies. Nucleic Acids Res. 2025 Jul 22;53(13):gkaf637. https://doi.org/10.1093/nar/gkaf637