Introduction
Kikuchi-Fujimoto disease (KFD), or histiocytic necrotizing lymphadenitis, is a rare, benign, self-limiting condition first described in young Asian women but now known to affect all ethnicities and age groups.[1] It most commonly presents with cervical lymphadenopathy and fever, accounting for approximately 0.6% of all pathologically examined lymphadenopathies.[2] Other systemic symptoms may include malaise, weight loss, arthralgia, and skin rashes.[3] The etiology remains uncertain, but infectious and autoimmune triggers in genetically predisposed individuals are suspected. Various bacterial and viral agents, such as Brucella, Bartonella, Toxoplasma, Mycobacterium species, and especially Epstein-Barr virus (EBV), have been implicated.[4,5] A genetic susceptibility involving HLA-DPA1 and HLA-DPB1 alleles has been reported, particularly in Asian populations.[6] KFD has also been linked to autoimmune disorders, notably systemic lupus erythematosus (SLE).[7,8]KFD is self-remitting but must be distinguished from serious infections, autoimmune diseases, and malignancies, particularly lymphoma and tuberculosis.[7-9] Diagnosis is confirmed by lymph node biopsy showing histiocytic necrotizing lymphadenitis without neutrophil infiltration.[7] The disease typically resolves within months, with recurrence rates of 3–4%.[8,10] Management is supportive, while corticosteroids or immunosuppressants are reserved for severe or recurrent cases.[10]
Hemophagocytic lymphohistiocytosis (HLH), by contrast, is a life-threatening hyperinflammatory syndrome caused by persistent activation of macrophages and cytotoxic lymphocytes, resulting in multiorgan damage. HLH may be primary (genetic) or secondary to infections, malignancy, or autoimmune diseases.[11,12] Diagnostic criteria include fever, splenomegaly, cytopenias, hypertriglyceridemia and/or hypofibrinogenemia, elevated ferritin, increased soluble IL-2 receptor, low NK-cell activity, and hemophagocytosis on tissue biopsy.[14] HLH treatment involves high-dose corticosteroids, etoposide, cyclosporine, and intravenous immunoglobulin (IVIG), with bone marrow transplantation indicated for refractory or familial disease.[15,16]
We describe a young woman who presented with lymphadenopathy and fever and was diagnosed with KFD complicated by HLH, illustrating the diagnostic and therapeutic challenges of this rare overlap.
Case Presentation
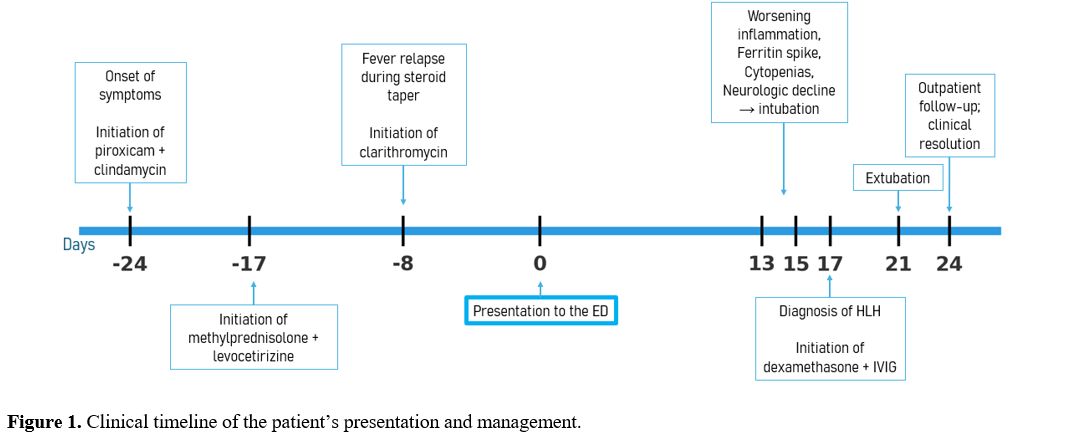 |
|
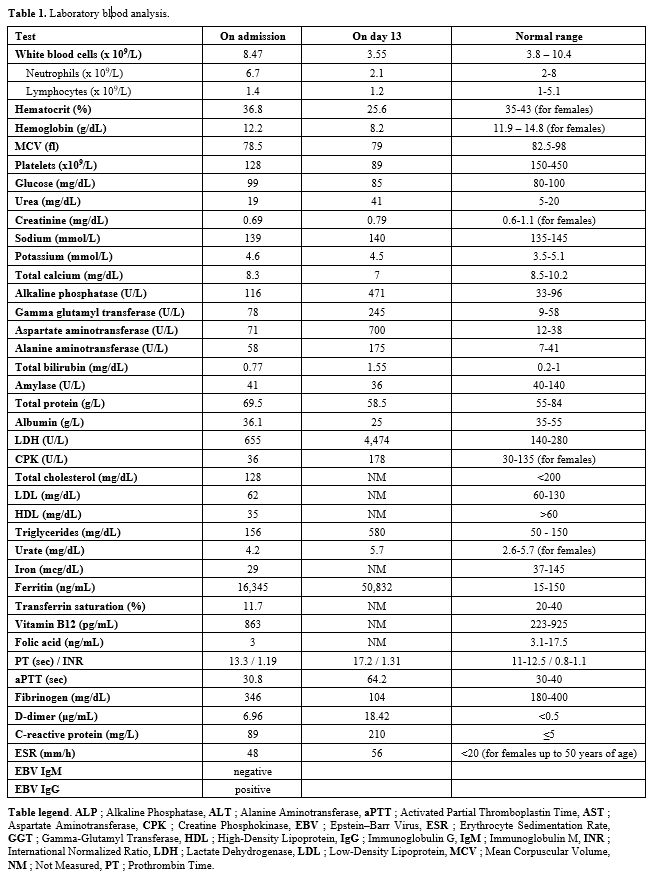
On admission, she was febrile (38.2°C), with mild hepatosplenomegaly and cervical and axillary lymphadenopathy. No rash or pharyngitis was present. Laboratory results (Table 1) showed mild thrombocytopenia, elevated lactate dehydrogenase, and hyperferritinemia. Inflammatory markers were high, while blood cultures and serologic tests for EBV (IgM negative, IgG positive, low viral load by PCR), Cytomegalovirus (CMV), Toxoplasmosis, Hepatitis B/C, and HIV were negative. Autoimmune testing, including ANA, anti-dsDNA, ANCA, and complement levels, was unremarkable. Imaging revealed hepatosplenomegaly (17 cm and 13 cm, respectively) and bilateral cervical and axillary lymphadenopathy.
 |
|
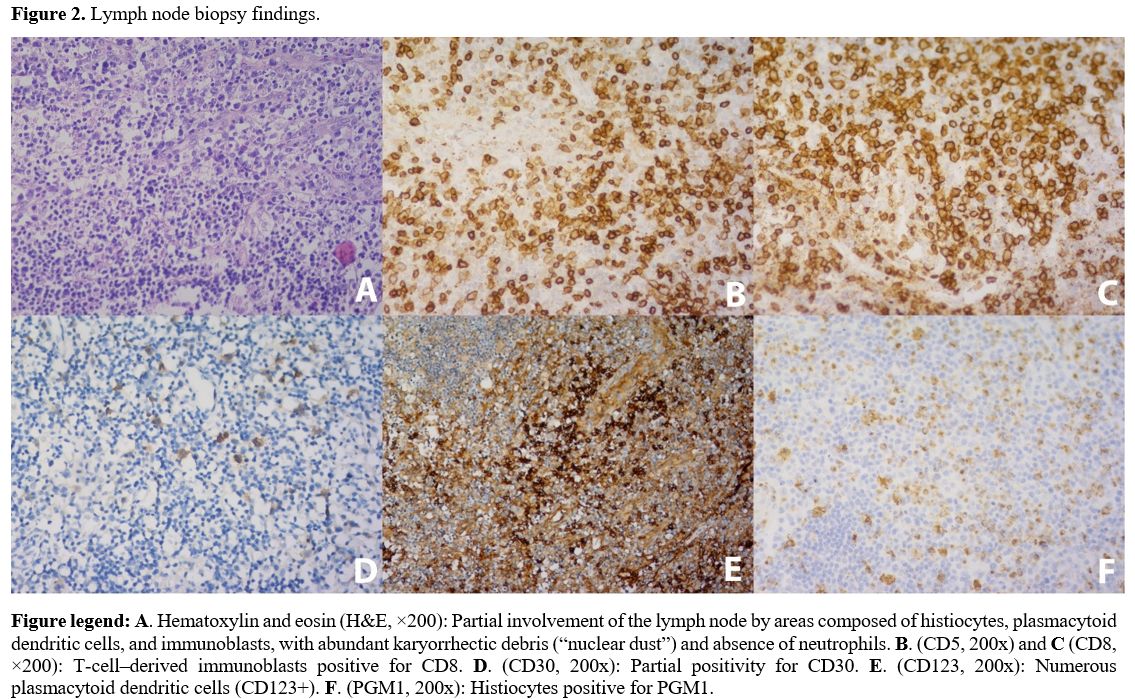
Empiric ceftriaxone followed by cefepime failed to improve symptoms. Cerebrospinal fluid (CSF) analysis revealed lymphocytic pleocytosis and mild protein elevation, but no evidence of infection. Our patient’s lymph node biopsy (Figure 2) demonstrates classic histopathological features of Kikuchi–Fujimoto disease (KFD). The areas of necrosis showed irregular, patchy, and often paracortical coagulative necrosis with abundant karyorrhectic debris, a hallmark pattern that helps distinguish KFD from other necrotizing lymphadenitis. The necrotic zones were populated by numerous CD8⁺ T-cell immunoblasts — a characteristic finding reflecting the cytotoxic T-cell–mediated immune response believed to drive KFD pathogenesis. Equally important was the prominence of CD123⁺ plasmacytoid dendritic cells. The absence of neutrophils and eosinophils is a critical morphologic discriminator; their presence would favor alternative diagnoses, such as suppurative lymphadenitis or autoimmune lymphadenitis. Histiocytes expressing PGM1 were abundant at the periphery of necrotic foci, forming the typical “crescentic” histiocytic pattern described in KFD. In contrast to lupus lymphadenitis, KFD lacks hematoxylin bodies, plasma cell–rich infiltrates, and immune complex deposition. The immunoblastic proliferation, while sometimes intense, remains polyclonal and accompanied by the characteristic necrotic pattern — findings that help distinguish KFD from T-cell lymphomas. The patchy necrosis, mixed inflammatory background, and absence of overt atypia in our case strongly favored KFD. On day 13, her condition deteriorated with persistent high-grade fevers (up to 39.6°C), cytopenias, elevated liver enzymes, LDH, and ferritin (Table 1). Neurological symptoms (confusion and seizures) developed, requiring intubation and intensive care. The repeated bone marrow biopsy again lacked hemophagocytosis, but her laboratory profile provided strong objective support for HLH. Using the HLH-2004 framework, she fulfilled five of eight diagnostic criteria (fever, splenomegaly, cytopenias in ≥2 lineages, hypertriglyceridemia/hypofibrinogenemia, and ferritin ≥500 µg/L), which by definition establishes the diagnosis in the absence of a confirmatory genetic mutation. The calculated H-score of 284 places her in the >99% probability range for reactive HLH by the validated Fardet H-score, further corroborating the clinical diagnosis and supporting urgent immunomodulatory treatment. She was treated with intravenous dexamethasone (20 mg/day) and IVIG (0.4 g/kg/day for 4 days). Fever and laboratory abnormalities improved markedly within one week, allowing extubation and transfer to the medical ward. She was discharged on a steroid taper and remained asymptomatic at 20-day follow-up, with normalization of laboratory values and resolution of lymphadenopathy. Although HLH-2004 recommends an etoposide–dexamethasone backbone for many patients, literature recognizes that in secondary (reactive) adult HLH, a short, aggressive trial of high-dose corticosteroids and/or IVIG is an accepted initial strategy because it can rapidly control the cytokine storm, avoiding the risks of cytotoxic therapy.[17,18] Since our patient showed significant clinical and laboratory improvement within days of starting dexamethasone and IVIG, we chose to continue this less-toxic regimen rather than immediately escalating to etoposide, which is reserved for persistent or progressive CNS disease.[17] In our patient, neurologic symptoms quickly reversed with systemic dexamethasone and IVIG, eliminating the immediate need for intrathecal therapy or upfront etoposide.
 |
|
Discussion
This case demonstrates an uncommon and severe presentation of KFD complicated by HLH. The patient’s constellation of fever, lymphadenopathy, and extreme hyperferritinemia initially suggested malignancy or infection, but histopathology confirmed KFD. HLH was subsequently diagnosed based on clinical and laboratory features, despite the absence of hemophagocytosis on biopsy. This finding is nonspecific and often absent early in the disease.[19]The overlapping clinical profiles of KFD and HLH — fever, cytopenias, lymphadenopathy, hepatosplenomegaly — make differentiation challenging. HLH, however, carries a far worse prognosis and demands urgent recognition and immunosuppressive therapy.[20] In our case, prompt administration of corticosteroids and IVIG likely prevented fatal progression.
Central nervous system (CNS) involvement in HLH occurs in up to 70% of cases and may manifest as seizures, encephalopathy, or altered consciousness, even with normal neuroimaging.[21] Our patient’s CSF lymphocytic pleocytosis and seizures indicated CNS involvement, which improved following immunomodulatory therapy.
The pathophysiological link between KFD and HLH involves the hyperactivation of CD8⁺ T cells and histiocytes, suggesting a shared immune dysregulation. In KFD, an aberrant or exaggerated CD8⁺ T-cell response — often in reaction to a viral or autoantigenic trigger — leads to apoptosis and formation of the characteristic necrotizing lymphadenitis.[4-8] When this process becomes systemically dysregulated, failure of immune homeostasis can lead to uncontrolled macrophage activation, cytokine storm, and the clinical picture of HLH (Supplementary materials). The biopsy in our case, which demonstrated abundant CD8⁺ immunoblasts and plasmacytoid dendritic cells, supports this mechanistic continuum. HLH superimposed on KFD, therefore likely represents an extreme point on the same immunologic spectrum rather than a distinct process.[22-23]
Regarding underlying triggers in our patient, although there was no evidence of acute EBV infection, positive IgG and low EBV viral load make a latent or past infection likely, which may have contributed. Corticosteroid exposure before presentation may also have partially suppressed early inflammatory signals while permitting progression of the underlying immune dysregulation; steroid tapering is a recognized trigger for rebound cytokine activation, which may have amplified the transition from KFD to overt HLH. Her obesity may also have played a role.[23] A literature review was conducted to identify published cases of HLH in association with KFD. A systematic search was performed using the terms “Kikuchi-Fujimoto”, “KFD”, AND “hemophagocytic lymphohistiocytosis”, “HLH”. The PubMed/MEDLINE and Google Scholar databases were searched. Exclusion criteria included studies lacking confirmation of KFD or HLH, review articles, and conference abstracts. A total of 35 unique cases (Supplementary material) meeting the inclusion criteria were identified. The following information was extracted: age, sex, presenting symptoms, laboratory findings, associated diagnoses, treatments administered, and clinical outcomes.
The median patient age was 20.5 years, with a slight male predominance, which contradicts the knowledge that KFD alone occurs more frequently in females. Cervical lymphadenopathy was the most common presentation (91%), and hyperferritinemia and elevated LDH were nearly universal. An additional unrelated diagnosis was made in 21 out of the 35 total patients, such as infection or systemic disorders, for example, SLE, juvenile myelomonocytic leukemia, and pregnancy. Corticosteroids were used in 83% of patients, while additional therapies (IVIG, etoposide, cyclosporine) were administered in 34%. If a secondary process was identified as a potential trigger for the intense inflammatory process, this was specifically targeted, through antivirals or antibiotics in cases of infection, antineoplastic agents (pralatrexate and bexarotene) in a patient with peripheral T-cell lymphoma and termination of pregnancy was considered in a female patient that experienced a spontaneous abortion. Despite treatment, mortality was 11%, highlighting the aggressive nature of this overlap syndrome.[17] KFD typically resolves spontaneously, but when complicated by HLH, aggressive immunosuppressive therapy is essential. IVIG and corticosteroids are considered first-line; refractory cases may require etoposide or cyclosporine.[15]
Conclusions
KFD is a rare, benign condition that may occasionally trigger secondary HLH, a potentially fatal hyperinflammatory state. Our patient’s presentation is largely consistent with patterns observed in our literature review. She was a young adult, consistent with the median age of 20.5 years reported in the literature. Her cervical lymphadenopathy, elevated LDH, and marked hyperferritinemia mirrored the most common reported features. Her severe neurologic involvement — encephalopathy and seizures — places her among the rarer and serious cases, as neurological symptoms are rarely seen in patients with fulminant disease. Similar to several published cases, she fulfilled HLH criteria despite the absence of hemophagocytosis on bone marrow biopsy, emphasizing that diagnosis should rely on clinical and laboratory criteria rather than histopathology alone.Clinicians should suspect HLH in KFD patients with persistent fever, cytopenias, or extreme hyperferritinemia. Diagnosis requires integration of clinical findings, laboratory results, and histopathology. Our literature review confirmed that patients with KFD-associated HLH usually exhibit extremely elevated serum ferritin and lactate dehydrogenase levels compared with patients with KFD alone. Prompt immunosuppressive treatment with corticosteroids and/or IVIG is critical for favorable outcomes. CNS involvement, although uncommon, should be anticipated in deteriorating patients. Symptoms include altered mental status, seizures, and loss of consciousness. Awareness of this rare but severe association is vital for timely recognition and management.
List of Abbreviations
• EBV – Epstein–Barr Virus
• HLH – Hemophagocytic Lymphohistiocytosis
• IVIG – Intravenous Immunoglobulin
• SLE – Systemic Lupus Erythematosus
• ANA – Antinuclear Antibody
• ANCA – Antineutrophil Cytoplasmic Antibody
• BMI – Body Mass Index
• CMV – Cytomegalovirus
• CSF – Cerebrospinal Fluid
• LDH – Lactate Dehydrogenase
• PCR – Polymerase Chain Reaction
• IgM – Immunoglobulin M
• IgG – Immunoglobulin G
• IL-2 – Interleukin-2
• H-score – Hemophagocytic Syndrome Probability Score
• NK-cell – Natural Killer Cell
• CNS – Central Nervous System
• AST – Aspartate Aminotransferase
• CPK – Creatine Phosphokinase
• ESR – Erythrocyte Sedimentation Rate
• GGT – Gamma-Glutamyl Transferase
• HDL – High-Density Lipoprotein
• LDL – Low-Density Lipoprotein,
• aPTT – Activated Partial Thromboplastin Time
• PT –Prothrombin Time
• INR –International Normalized Ratio
• F –Female
Author Contributions
MM: Conceptualization, Methodology, Data Curation, Investigation, Formal Analysis, Writing – Original Draft. PD: Supervision, Validation, Writing – Review & Editing. AH: Data Curation, Investigation. SL: Data Curation, Investigation. MM: Supervision, Review & Editing.Data Availability Statement
The data that support the findings of this study are available in the literature (See references section).References
- Bosch X, Guilabert A, Miquel R,
Campo E. Enigmatic
Kikuchi-Fujimoto disease: a comprehensive review. Am J Clin Pathol.
2004;122(1):141-152. https://doi.org/10.1309/YF081L4TKYWVYVPQ
PMid:15272543
- Kucukardali Y et al. Kikuchi-Fujimoto Disease:
analysis of 244 cases. Clin Rheumatol. 2007;26(1):50-54. https://doi.org/10.1007/s10067-006-0230-5
PMid:16538388
- Perry AM, Choi SM. Kikuchi-Fujimoto disease: a
review. Arch Pathol Lab Med. 2018;142(11):1341-1346. https://doi.org/10.5858/arpa.2018-0219-RA
PMid:30407860
- Hudnall
SD et al. Detection of human herpesvirus DNA in Kikuchi-Fujimoto
disease and reactive lymphoid hyperplasia. Mod Pathol.
2008;21(5):593-599.
- Lin HC et al. Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis in adults. Front Immunol. 2021;12:620472.
- Chiu CF et al. Genetic association of HLA class II alleles with Kikuchi disease. Arthritis Rheum. 2004;50(11):3641-3645.
- Dorfman RF, Berry GJ. Kikuchi's histiocytic necrotizing lymphadenitis: an analysis of 108 cases. Am J Surg Pathol. 1988;12(11):847-858.
- Liu Y et al. Clinical and laboratory features of Kikuchi-Fujimoto disease and its association with autoimmune disorders. Front Med. 2022;9:812481
- Kwon
SY, Kim TK. Sonographic findings of Kikuchi disease: differentiation
from tuberculous lymphadenitis. J Ultrasound Med. 2017;36(9):1913-1922.
- Kim
JH et al. Clinical outcome and recurrence of Kikuchi-Fujimoto disease.
Eur J Intern Med. 2019;64:68-73.
- Ramos-Casals
M et al. Adult haemophagocytic syndrome. Lancet.
2014;383(9927):1503-1516. https://doi.org/10.1016/S0140-6736(13)61048-X
PMid:24290661
- Brisse
E et al. Pathophysiology of hemophagocytic lymphohistiocytosis (HLH):
mechanisms and clinical implications. J Allergy Clin Immunol.
2016;138(4):993-1008.
- Henter
JI et al. HLH-2004 diagnostic and therapeutic guidelines. Pediatr Blood
Cancer. 2007;48(2):124-131. https://doi.org/10.1002/pbc.21039
PMid:16937360
- Jordan
MB et al. How I treat hemophagocytic lymphohistiocytosis. Blood.
2011;118(15):4041-4052. https://doi.org/10.1182/blood-2011-03-278127
PMid:21828139 PMCid:PMC3204727
- Imashuku
S. Clinical features and treatment strategies of Epstein-Barr
virus-associated HLH. Crit Rev Oncol Hematol. 2002;44(3):259-272. https://doi.org/10.1016/S1040-8428(02)00117-8
PMid:12467966
- Henter
JI et al. HLH-2004: Diagnostic and therapeutic guidelines for
hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007
Feb;48(2):124-31. https://doi.org/10.1002/pbc.21039
PMid:16937360
- Zoref-Lorenz
A et al. Recognizing and Managing Secondary Hemophagocytic
Lymphohistiocytosis in Adults: A Practical Clinical Guide. Hematol
Oncol Clin North Am. 2025 Jun;39(3):577-596. https://doi.org/10.1016/j.hoc.2025.02.007
PMid:40222878
- Lee
KY et al. Hemophagocytic lymphohistiocytosis associated with Kikuchi
disease: literature review and analysis of 35 cases. Clin Rheumatol.
2015;34(4):691-697.
- Rivière
S et al. Reactive hemophagocytic syndrome in adults: a retrospective
analysis of 162 patients. Am J Med. 2014;127(11):1118-1125. https://doi.org/10.1016/j.amjmed.2014.04.034
PMid:24835040
- Horne
A et al. Central nervous system involvement in HLH: a prospective
study. Blood. 2008;112(3):843-849.
- Gioia,
C et al. Pathogenesis of Hemophagocytic Lymphohistiocytosis/Macrophage
Activation Syndrome: A Case Report and Review of the Literature. Int.
J. Mol. Sci. 2024, 25, 5921. https://doi.org/10.3390/ijms25115921
PMid:38892108 PMCid:PMC11173133
- Wu,
Y. et al. Hemophagocytic lymphohistiocytosis: current treatment
advances, emerging targeted therapy and underlying mechanisms. J
Hematol Oncol 17, 106 (2024). https://doi.org/10.1186/s13045-024-01621-x
PMid:39511607 PMCid:PMC11542428
- Obeagu
EI et al. Body mass index and hemophagocytic lymphohistiocytosis: a
risk assessment in HIV-positive leukemia patients. Ann Med Surg (Lond).
2025 Mar 28;87(6):3424-3434. https://doi.org/10.1097/MS9.0000000000003195
PMid:40486594 PMCid:PMC12140742
Supplementary Files
 |
Supplementary Table 1. References |
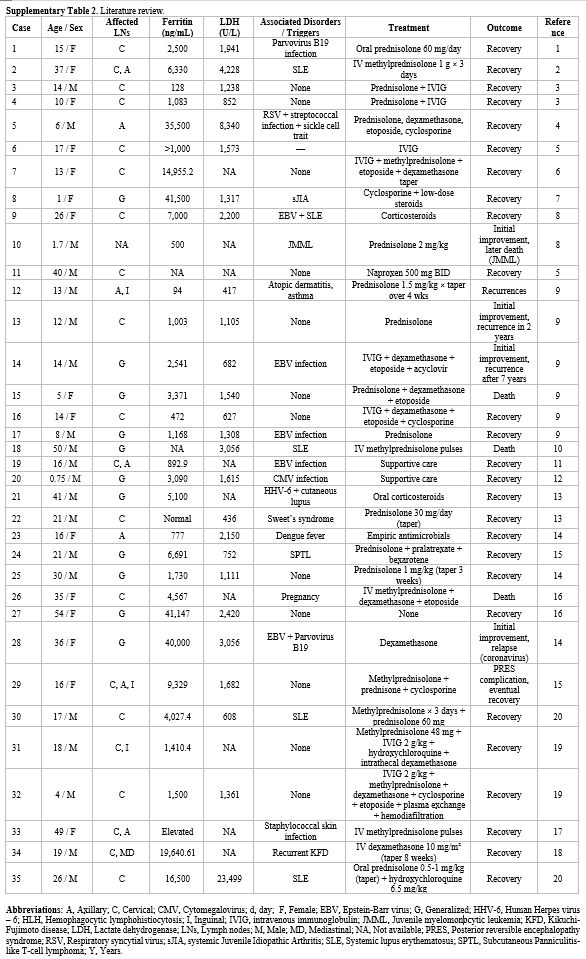 |
Supplementary Table 2.
Literature review. |
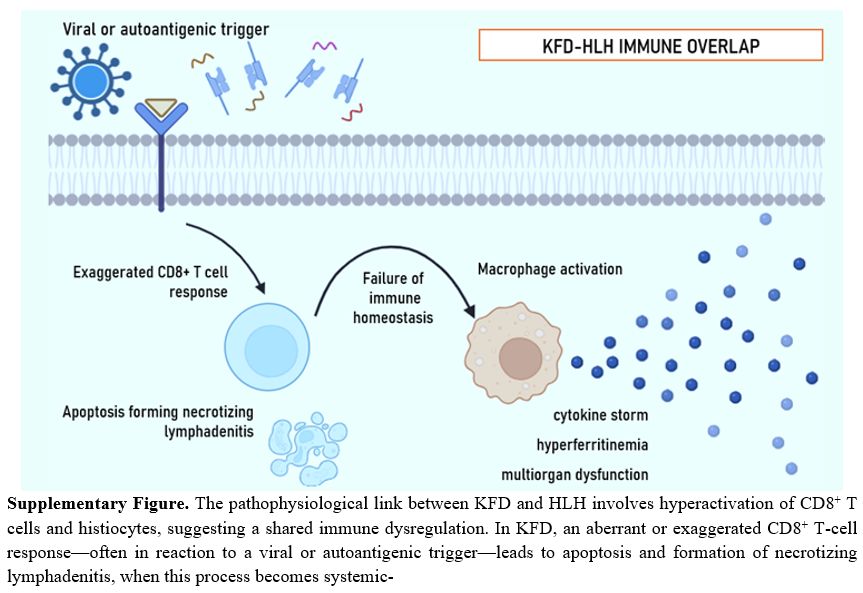 |
Supplementary Figure.
The
pathophysiological link between KFD and HLH involves hyperactivation of
CD8⁺ T cells and histiocytes, suggesting a shared immune dysregulation.
In KFD, an aberrant or exaggerated CD8⁺ T-cell response—often in
reaction to a viral or autoantigenic trigger—leads to apoptosis and
formation of necrotizing lymphadenitis, when this process becomes
systemic |