Introduction
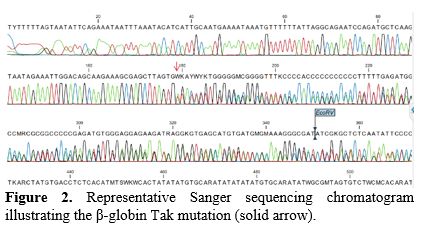
Hemoglobin Tak (Hb Tak) represents an uncommon hemoglobin variant with elevated oxygen affinity, arising from the addition of two nucleotides (AC) at the termination codon of the β-globin gene, affecting the region between codons 146 and 147, resulting in a frameshift at codon 147 and elongation of the β-globin chain by 11 amino acids.[1] This structural alteration increases oxygen affinity and clinically manifests as secondary erythrocytosis.[2,3] Secondary polycythemia has been documented not only among individuals with Hb Tak/β-thalassemia but also across other Hb Tak- related genotypes, such as homozygous Hb Tak and compound heterozygous Hb Tak/HbE.[4,5] Only several Hb Tak cases have been reported worldwide, mostly as isolated case reports, with the first case reported in Thailand.[6]No cases have been documented in Vietnam to date. At the Thalassemia Center of the National Institute of Hematology and Blood Transfusion, the largest center in Vietnam for screening, diagnosis, and treatment of hemoglobin disorders, we identified and followed 4 patients with Hb Tak/β-thalassemia. By reporting this case series, we aim to expand the existing literature and highlight the diagnostic importance of hemoglobin analysis in patients presenting with erythrocytosis accompanied by microcytic, hypochromic red blood cells.
We have described four patients diagnosed with heterozygous Hb Tak/β-thalassemia at our center between 2014 and 2025. Case identification was based on hemoglobin profiling and molecular genetic analyses. Clinical and laboratory information was obtained from medical records. Hematological indices were measured using automated analyzers. Hemoglobin fractions were evaluated through HPLC. Molecular analyses included PCR-based assays to screen for the JAK2V617F mutation, ruling out PV. In addition, β- and α-thalassemia mutations were examined using established PCR techniques, and the presence of Hb Tak was verified by Sanger sequencing targeting the β-globin gene.
The size of the liver and spleen was measured using ultrasonography. Splenomegaly was defined by either a palpable spleen on abdominal examination or a splenic length greater than 12 cm on ultrasonography.[7] The severity of splenic enlargement was classified according to the Hackett grading system.[8]
Case Series
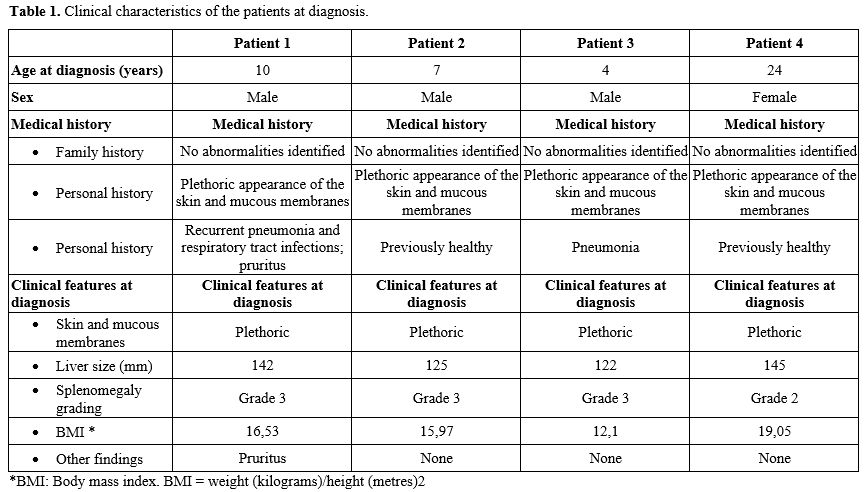
The clinical features:Patient 1: A 10-year-old boy presented with a medical history notable for recurrent episodes of pneumonia beginning at 5 months of age. Since the age of 15 months, he had exhibited a plethoric appearance, affecting both the skin and mucous membranes. By the age of 10 years, the facial and mucosal plethora became progressively more pronounced, accompanied by frequent headaches, without motor weakness or paralysis. He was subsequently evaluated at our institution and diagnosed with polycythemia secondary to Hb Tak/β-thalassemia.
Patient 2: A 7-year-old male patient exhibited a plethoric appearance of the skin and ocular mucosa since birth. Despite this finding, the patient demonstrated normal physical growth and development, as evidenced by weight and height; therefore, no medical evaluation or treatment was sought during early childhood. At the age of seven, the patient developed bilateral ocular swelling and pain accompanied by conjunctival congestion. Initial assessment at a regional healthcare facility revealed an elevated hemoglobin level. The patient was subsequently referred to our hospital, where he was diagnosed with secondary polycythemia associated with Hb Tak/β-thalassemia.
Patient 3: A 6-year-old male patient had a history of plethoric appearance of the skin and mucous membranes since the age of three. At six years of age, he was presented with fever, cough, and dyspnea and was admitted to our hospital. He was diagnosed with pneumonia, pulmonary arterial hypertension, and polycythemia associated with Hb Tak/β-thalassemia. The patient subsequently received antibiotic therapy and therapeutic phlebotomy.
Patient 4: A 24-year-old female patient had a long-standing history of plethoric skin and mucous membrane appearance, which had not been previously investigated. She married at the age of 23 and delivered her first child via spontaneous vaginal delivery at 24 years of age. One week postpartum, she developed severe, diffuse abdominal pain accompanied by nausea and recurrent vomiting, without fever. She was diagnosed with acute mesenteric ischemia secondary to postpartum mesenteric thrombosis in the setting of secondary polycythemia due to Hb Tak/β-thalassemia.
The clinical characteristics of patients were summarized in Table 1.
 |
|
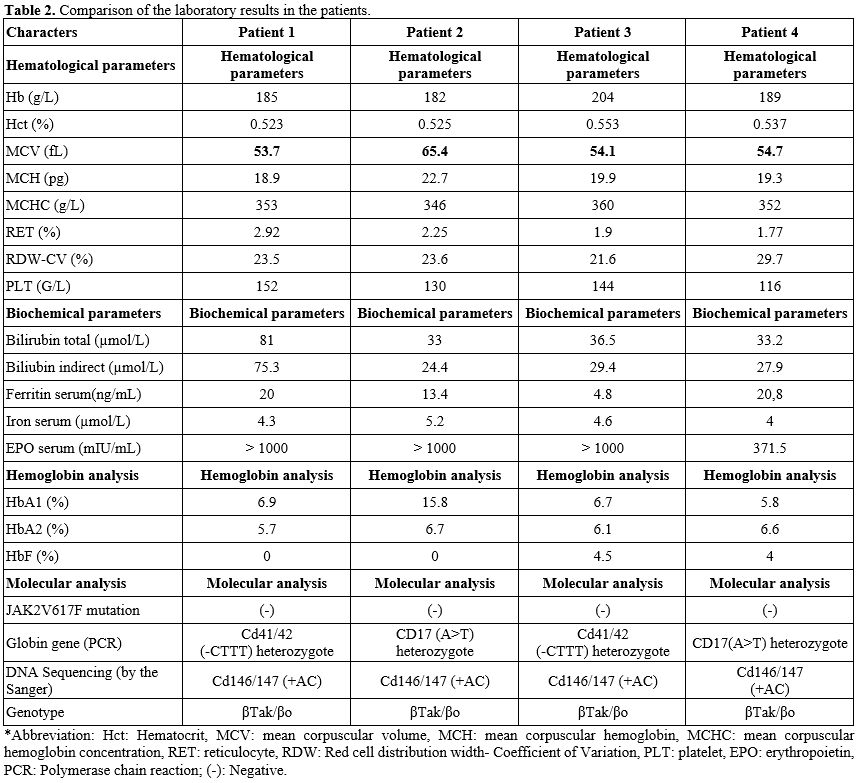
Laboratory features at diagnosis. All four patients demonstrated erythrocytosis, with hemoglobin levels exceeding 160 g/L in the female patient and 165 g/L in the male patients, and elevated hematocrit values greater than 0.50 L/L. Peripheral blood smear examination revealed microcytic, hypochromic red blood cells with marked anisocytosis and poikilocytosis, characterized by multiple abnormal morphologies, including target cells, teardrop cells, ovalocytes, and others. Reticulocyte counts were increased in all cases, indicating enhanced erythropoietic activity (Table 2).
 |
|
Furthermore, all patients exhibited laboratory evidence of ongoing hemolysis, as reflected by elevated indirect bilirubin levels. Reduced serum ferritin concentrations (depleted iron stores) and markedly elevated erythropoietin levels were also consistently observed across all four cases (Table 2).
Hemoglobin electrophoresis: all four cases showed increased HbA2 and the presence of another hemoglobin type (suspected to be Hb Tak) in very high proportions, ranging from 76.6% to 83.6% (Table 2). Only cases 3 and 4 showed HbF (Table 2 and Figure 1).
 |
|
In the entire case series, they were tested for the JAK2V617F gene mutation, and all results were negative. The globin gene mutation was identified in all cases with a genotype of βTak/βo (Table 2 and Figure 2).
 |
|
Discussion
Hemoglobin Tak (Hb Tak) is one of several very rare high-oxygen-affinity β-globin variants, first described in Thailand in 1971.[6,9,10] To date, while recent literature has begun to report case series of high-oxygen-affinity hemoglobin variants in general,[11] data on heterozygous Hb Tak/β-thalassemia have been dominated by isolated case reports, with only a few small case series reported, notably by Panyasai et al. and Rukwong et al.,[12,13] all originating from Thailand. In this context, our report represents the first cluster of Hb Tak/β-thalassemia cases described in Vietnam. This case series adds to the currently limited body of evidence on this rare genotype and provides valuable data to improve diagnostic awareness, particularly in patients presenting with atypical thalassemia phenotypes.Clinically, the patients showed facial plethora, pinkish mucous membranes, mild to moderate hepatosplenomegaly, findings comparable to those reported in other high-oxygen-affinity hemoglobin variants (Table 1). Hematologically, all cases displayed microcytic, hypochromic indices with elevated HbA₂ (>3.5%)[14] and/or HbF, consistent with β-thalassemia carrier status.[15] Notably, the abnormal hemoglobin fraction accounted for a high proportion (76.6%-83.6%) on HPLC. This predominance of Hb Tak is likely attributable to the absence of normal β-globin chain synthesis in β-thalassemia, resulting in Hb Tak becoming the nearly dominant hemoglobin species. Molecular analysis confirmed heterozygous β-thalassemia mutations in all patients, and Sanger sequencing verified the characteristic AC insertion between codons 146 and 147, which is responsible for Hb Tak (Figure 2).
Despite elevated hemoglobin levels, reticulocyte counts were also increased, indicating marked erythroid hyperplasia in bone marrow. This finding is consistent with the pathophysiology of Hb Tak/β-thalassemia: the markedly increased oxygen affinity of Hb Tak impairs oxygen delivery to tissues, leading to relative tissue hypoxia. This, in turn, stimulates renal EPO production and compensatory erythropoiesis. Accordingly, serum EPO levels were markedly elevated in all patients; the combination with negative JAK2V617F mutation testing allowed PV to be reasonably excluded without the need for invasive bone marrow biopsy.[16]
Similar to thalassemia patients in general, individuals in our cohort exhibited evidence of chronic hemolysis, as reflected by elevated total bilirubin levels, predominantly unconjugated (Table 2). However, in contrast to the iron overload commonly observed in thalassemia, patients with Hb Tak/β-thalassemia in our series showed reduced iron stores (Table 2). This finding may be attributed to increased iron utilization driven by sustained erythroid hyperplasia in response to tissue hypoxia. When heightened erythropoietic demand persists in a setting of inadequate iron supply, patients may develop functional or absolute iron deficiency.
Concerning potential complications, the first and third patients in our series experienced recurrent episodes of pneumonia. This manifestation has been previously reported in a pediatric case from Thailand with homozygous Hb Tak, in whom polycythemia and increased blood viscosity were considered contributing factors.[4] In addition, the fourth patient developed mesenteric thrombosis, representing a thrombotic complication in our cohort. Notably, high-oxygen-affinity hemoglobin variants have occasionally been reported to be associated with thrombotic or hemorrhagic events, including recurrent intracranial hemorrhage in patients with Hb Tak.[17] Taken together, our observations, in comparison with previously reported hemorrhagic and thrombotic cases, suggest that patients with Hb Tak/β-thalassemia may have a predisposition to vascular complications. Such complications may become clinically relevant under certain conditions, including dehydration, infection, pregnancy, or increased tissue oxygen demand.[18]
About treatment and clinical management, due to limited available data, standardized management guidelines for these patients are lacking. In our center, low-dose aspirin (81 mg) was considered based on clinical symptoms and hematocrit levels to mitigate thrombotic risk, and phlebotomy was performed every 6-8 weeks to maintain hematocrit below 45%.
Conclusions
Our case series expands current knowledge of Hb Tak/β-thalassemia, an extremely rare genotype associated with a distinctive yet frequently overlooked phenotype. The coexistence of erythrocytosis and microcytosis should raise suspicion of high-oxygen-affinity hemoglobin variants rather than attributing it solely to iron deficiency or primary erythrocytosis. Importantly, in pediatric patients presenting with erythrocytosis, this group of hemoglobin disorders should be actively considered during the diagnostic workup. Incorporation of hemoglobin analysis and targeted molecular testing into the diagnostic algorithm for patients (particularly children) with unexplained erythrocytosis and microcytosis may help prevent unnecessary invasive investigations and inappropriate treatments, while facilitating timely diagnosis and appropriate genetic counseling for affected families.Author contributions
Thi Chi Nguyen and Ngoc Dung Nguyen: Conceptualization, Data curation, Writing – Original draft. Thi Nguyet Anh Phi and Thi Thu Ha Nguyen: Supervision, Literature review, Writing – Review & editing. Xuan Hai Le and Duc Luong Vu: Investigation, Data collection.References
- M.-C. Shih, K.-H. Wu, S.-C. Liu, and J.-G. Chang,
"Hb Tak: a beta chain elongation at the end of the beta chain, in a
Taiwanese," Hemoglobin, vol. 29, no. 1, pp. 65-67, 2005. https://doi.org/10.1081/HEM-47635 PMid:15768557
- P.
Charoenkwan, P. Thanarattanakorn, S. Chaovaluksakul, S.
Sittipreechacharn, R. Sae-Tang, and T. Sanguansermsri, "HEMATOLOGICAL
AND MOLECULAR CHARACTERIZATION OF BETA-THALASSEMIA/HB TAK COMPOUND
HETEROZYGOTE," SOUTHEAST ASIAN J TROP MED PUBLIC HEALTH, vol. 34, no.
2, 2003.
- S.
Panyasai, S. Sakkhachornphop, and S. Pornprasert, "Diagnosis of
Compound Heterozygous Hb Tak/β-Thalassemia and HbD-Punjab/β-Thalassemia
by HbA2 Levels on Capillary Electrophoresis," Indian J Hematol Blood
Transfus, vol. 34, no. 1, pp. 110-114, Jan. 2018. https://doi.org/10.1007/s12288-017-0810-3 PMid:29398808 PMCid:PMC5786607
- V.
S. Tanphaichitr, V. Viprakasit, G. Veerakul, K. Sanpakit, and P.
Tientadakul, "Homozygous hemoglobin Tak causes symptomatic secondary
polycythemia in a Thai boy," J Pediatr Hematol Oncol, vol. 25, no. 3,
pp. 261-265, Mar. 2003. https://doi.org/10.1097/00043426-200303000-00016 PMid:12621249
- N.
Teawtrakul, C. Sirijirachai, G. Chansung, and G. Fucharoen, "Compound
heterozygous Hb Tak/Hb E causes secondary erythrocytosis in a Thai
family," Hemoglobin, vol. 34, no. 2, pp. 165-168, Jan. 2010. https://doi.org/10.3109/03630261003680498 PMid:20353353
- G.
Flatz, J. L. Kinderlerer, J. V. Kilmartin, and H. Lehmann, "Haemoglobin
Tak: a variant with additional residues at the end of the beta-chains,"
Lancet, vol. 1, no. 7702, pp. 732-733, Apr. 1971. https://doi.org/10.1016/S0140-6736(71)91994-5 PMid:4101432
- J.
Chapman, A. Goyal, and A. M. Azevedo, "Splenomegaly," in StatPearls,
Treasure Island (FL): StatPearls Publishing, 2025. Accessed: Dec. 30,
2025. [Online]. Available: http://www.ncbi.nlm.nih.gov/books/NBK430907/
- V. S. Dabadghao and A. M. Raskar, "A Clinicohaematological Profile of Splenomegaly," vol. 54, no. 1, 2012.
- "Five
Hemoglobin Variants in a Double Heterozygote... : Acta Haematologica,"
Ovid. Accessed: Dec. 25, 2025. [Online]. Available: https://www.ovid.com/journals/achaah/fulltext/10.1159/000353123~five-hemoglobin-variants-in-a-double-heterozygote-for---and
- J.
Yudin and M. Verhovsek, "How we diagnose and manage altered oxygen
affinity hemoglobin variants," Am J Hematol, vol. 94, no. 5, pp.
597-603, May 2019. https://doi.org/10.1002/ajh.25425 PMid:30690774
- V.
Komninaka et al., "High-Oxygen-Affinity Hemoglobins-Case Series and
Review of the Literature," Journal of Clinical Medicine, vol. 13, no.
2, Jan. 2024. https://doi.org/10.3390/jcm13020458 PMid:38256595 PMCid:PMC10815990
- S.
Panyasai, S. Sakkhachornphop, and S. Pornprasert, "Diagnosis of
Compound Heterozygous Hb Tak/β-Thalassemia and HbD-Punjab/β-Thalassemia
by HbA2 Levels on Capillary Electrophoresis," Indian J Hematol Blood
Transfus, vol. 34, no. 1, pp. 110-114, Jan. 2018. https://doi.org/10.1007/s12288-017-0810-3 PMid:29398808 PMCid:PMC5786607
- P.
Rukwong et al., "Clinical and hematological characteristics of
beta-plus thalassemia and uncommon beta-chain hemoglobin variants in
Northern Thailand," Annals of Medicine, vol. 57, no. 1, p. 2551815,
Dec. 2025. https://doi.org/10.1080/07853890.2025.2551815 PMid:40888401 PMCid:PMC12404072
- "Tietz,
N.W. (1986) Textbook of Clinical Chemistry, W.B.Saunders Company,
Philadelphia. - References - Scientific Research Publishing." Accessed:
Jan. 05, 2026. [Online]. Available: https://www.scirp.org/reference/referencespapers?referenceid=24015
- "Prevention
and Diagnosis of Haemoglobinopathies: A Short Guide for Health
Professionals and Laboratory Scientists (2016)," TIF. Accessed: Dec.
30, 2025. [Online]. Available: https://thalassaemia.org.cy/publications/tif-publications/prevention-and-diagnosis-of-haemoglobinopathies-a-short-guide-for-health-professionals-and-laboratory-scientists/
- "American Journal of Hematology | Blood Research Journal | Wiley Online Library." Accessed: Dec. 25, 2025. [Online]. Available: https://onlinelibrary.wiley.com/doi/10.1002/ajh.27002
- B.
S. Venkataramany, S. Najib, T. Sheikh, A. Saste, and D. Oostra,
"Recurrent Intracranial Hemorrhages in a Patient With Hb Tak and Hb E,"
AIM Clinical Cases, vol. 3, no. 6, p. e230997, Jun. 2024. https://doi.org/10.7326/aimcc.2023.0997
- F.
Tongprasert, P. Charoenkwan, K. Srisupundit, and A. Tantiworawit,
"Secondary erythrocytosis caused by hemoglobin Tak/β0 -thalassaemia
disease during pregnancy: A case report," Journal of Obstetrics and
Gynaecology, pp. 1-2, Dec. 2016. https://doi.org/10.1080/01443615.2016.1249356 PMid:27966384