In this setting, laboratory trends must be interpreted within a sickle cell-specific pathophysiological framework. Disproportionate lactate dehydrogenase elevation, abrupt thrombocytopenia, marked erythroblastosis, or mismatch between inflammatory markers and clinical severity may signal mechanisms distinct from those typically encountered in the general population.[3]
These complexities highlight the importance of stepwise evaluation and early involvement of specialized hemoglobinopathy centres in case clinical evolution is atypical or rapid.[4]
We report the case of abrupt multiorgan deterioration in a young woman with haemoglobin sickle-beta-thalassemia (HbS/β⁺-thalassaemia) initially managed as an uncomplicated vaso-occlusive episode. The diagnostic process evolved through sequential reassessment of clinical and laboratory findings, illustrating how systematic differential reasoning can uncover an unexpected underlying mechanism. This case highlights the importance of maintaining a broad differential diagnosis and reassessing initial assumptions when the clinical course deviates from the anticipated trajectory.
Report of the case
Case presentation and clinical history. A 20-year-old woman with sickle cell/thalassemia presented to an emergency department with severe pain involving both upper and lower limbs. She was receiving hydroxyurea (20 mg/kg/day) and underwent occasional erythrocytapheresis prior to long-haul flights. Her clinical phenotype was predominantly haemolytic, with fewer VOCs controlled with home analgesia. She had no prior history of multiorgan complications. She presented with intense limb pain (VAS >8) unresponsive to home therapy, consistent with VOCs.Initial assessment and clinical evolution. Initial assessment was performed at a peripheral hospital without on-site haemoglobinopathy expertise. On examination, she was afebrile and hemodynamically stable, with normal oxygen saturation on room air. No cardiac, pulmonary, or abdominal abnormalities were detected. Initial laboratory tests showed: white blood cells (WBC) 6,700/μL, haemoglobin (Hb) 10.8 g/dL, Platelets 180,000/μL, D-dimer 1,000 ng/mL, LDH 300 U/L, CRP 0.68 mg/dL, HbS 50%, HbF 20%.
She was treated according to standard VOC management with morphine, intravenous hydration, low-flow oxygen, and prophylactic low-molecular-weight heparin.
Within 24 hours of symptom onset, she rapidly progressed to acute respiratory failure accompanied by confusion and fever, requiring intubation and admission to the intensive care unit (ICU) on day +2.
Repeat laboratory testing showed: WBC 4.58 x10^3/uL, Hb 9.3 g/dL, platelets 89 x10^3/uL, nucleated red blood cells (NRBC) 0.05 x10^3/uL (1.1/100 WBC), reticulocytes 0.128 x10^6/uL (3.96%), alanine aminotransferase (ALT) 36 U/L, aspartate aminotransferase (AST) 73 U/L, creatine kinase (CK) 211 U/L, LDH 1,488.8 U/L, total bilirubin 3.7 mg/dL (direct bilirubin 0.8 mg/dL), creatinine 0.54 mg/dL, undetectable haptoglobin, CRP 10.95 mg/dL, and procalcitonin 0.44 ng/mL. Additional laboratory data are reported in Tables 1A and 1B. Parvovirus B19 testing was negative. Brain computed tomography (CT) was unremarkable. CT pulmonary angiography excluded pulmonary embolism but shows bilateral pulmonary infiltrates. Transthoracic echocardiography showed right ventricular dilatation and dysfunction consistent with acute pressure overload.
 |
|
Differential diagnosis and diagnostic reassessment. At the referring ICU, the initial working diagnosis was septic deterioration secondary to pulmonary infection complicating VOC. Broad-spectrum antibiotic therapy with piperacillin/tazobactam was started, vasopressor support with noradrenaline was required, and an automated red cell exchange was performed. Due to ongoing clinical instability and the need for specialised expertise, the patient was transferred to our hemoglobinopathy referral centre. On arrival (day +4), laboratory findings showed rapidly worsening anaemia and thrombocytopenia with apparently normal WBC count and unaltered RBC indices (Table 1A-B). Tests showed evolving multiorgan dysfunction: LDH 2,482 U/L; CK 514 U/L; AST 26 U/L; ALT 127 U/L; gamma-GT 38 U/L; Alkaline phosphatase 137 U/L; total bilirubin 2.0 mg/dL and direct 0.58 mg/dL; ferritin 4,235 ng/mL; T-troponin 213 pg/mL; myoglobin 72 ng/mL; NT-proBNP 7,309 pg/mL.
The combination of progressive cytopenias, neurological impairment, and organ dysfunction raised concern for thrombotic microangiopathy, particularly thrombotic thrombocytopenic purpura (TTP). ADAMTS13 activity was reduced (33%) but remained above 10% and ADAMTS13 inhibitors were negative. Peripheral blood smear showed marked erythroblastosis without schistocytes.
Final diagnosis. The absence of microbiological evidence, together with the lack of sustained response to antimicrobial therapy, also made sepsis less convincing as the unifying diagnosis. Moreover, within the context of sickle cell disease, disease-specific complications were considered more plausible explanations for the rapidly progressive clinical picture. Despite initial concern for thrombotic microangiopathy, several elements were incongruent with this diagnosis. The absence of schistocytes on peripheral smear and ADAMTS13 activity above the critical threshold strongly argued against a diagnosis of classical thrombotic thrombocytopenic purpura. Moreover, the earliest biochemical abnormalities were characterised by disproportionate elevations in creatine kinase and lactate dehydrogenase, which preceded the development of overt cytopenias. The marked peripheral erythroblastosis suggested acute marrow disruption rather than primary microangiopathy.
The combination of rapidly progressive respiratory failure, neurological impairment with unremarkable brain CT imaging, evolving multiorgan dysfunction, and an erythroblastic blood picture prompted reconsideration of the diagnostic hypothesis.
Further investigations included MRI, bronchoalveolar lavage (BAL) cytology, and bone marrow biopsy.
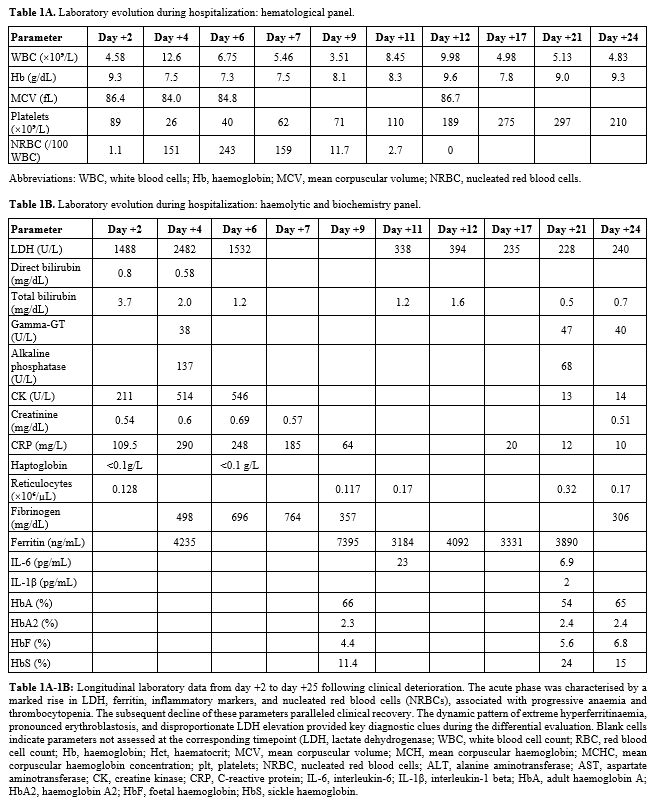
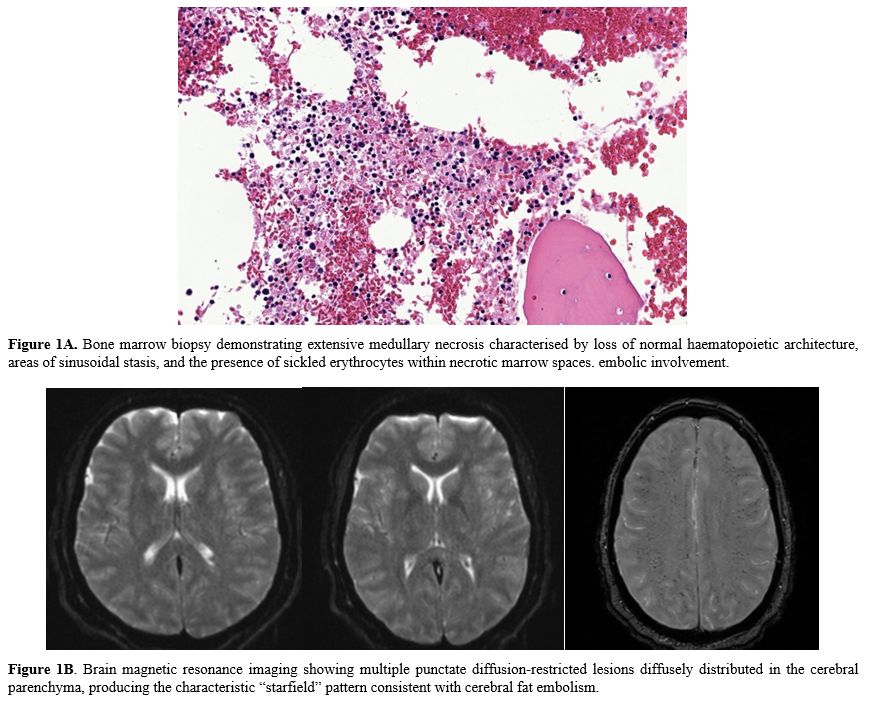
Brain imaging demonstrated a characteristic starfield pattern consistent with cerebral fat embolism, and BAL cytology identified fat globules. Bone marrow biopsy subsequently revealed extensive medullary necrosis with sinusoidal stasis and sickled erythrocytes (Figure 1).
 |
|
Taken together, the clinical, laboratory, radiological, and histopathological findings confirmed the diagnosis of acute bone marrow necrosis (BMN) leading to secondary fat embolism syndrome (FES).
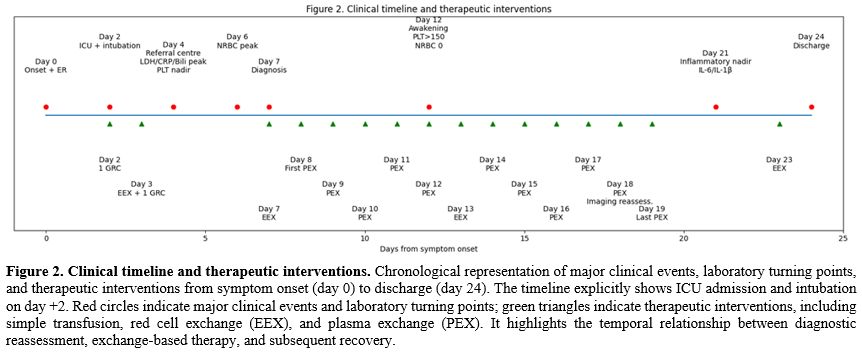
Treatment and outcome. Therapeutic plasma exchange (PEX) with plasma replacement was initiated on day +7. During her ICU stay, she underwent 11 PEX sessions and 2 red cell exchange procedures with supportive simple transfusions as clinically indicated (Figure 2).
 |
|
Clinical recovery was progressive. Haematological parameters and haemolytic indices improved by day +9. Despite the striking abnormalities on brain MRI, the patient regained consciousness by day +12 and achieved complete neurological recovery, without residual motor, sensory, or autonomic deficits. Follow-up laboratory tests showed normalisation of inflammatory markers and CK levels. Repeat brain MRI showed marked radiological improvement without structural sequelae. She was subsequently referred for structured neuro-rehabilitation and achieved full functional recovery.
Discussion
BMN encompasses a spectrum ranging from focal marrow infarction during VOC to extensive, life-threatening medullary destruction.[5] While localised marrow infarction is common and usually self-limited in SCD, the generalised variant is rare and may precipitate systemic FES and multiorgan dysfunction.[5] The syndrome remains under-recognised because its earliest manifestations overlap with more common complications such as infection and ACS().[6-7]Our patient's course was highly consistent with the phenotype described in published series of BMN/FES: a seemingly uncomplicated pain crisis followed by abrupt respiratory and neurological deterioration, a rapid fall in haemoglobin and platelets, and marked LDH elevation.[4,6-9]
A particularly informative clue was the striking erythroblastosis. In contrast to classical TTP, BMN/FEStypically produces a leukoerythroblastic picture with numerous nucleated red blood cells, whereas TTP is characterised by microangiopathic haemolysis with prominent schistocytosis and severe ADAMTS13 deficiency.[3] Therefore, although occasional schistocytes may be observed in critically ill patients, the predominance of erythroblastosis over microangiopathic fragmentation should favour marrow disruption rather than primary thrombotic microangiopathy.[2,8] This distinction proved critical in redirecting the diagnostic pathway: the main differential diagnoses considered are in Table 2.
 |
|
Marked hyperferritinaemia represented another useful clue. In BMN/FES, ferritin elevation probably reflects the combined effect of tissue necrosis, cytokine-driven acute-phase activation, and secondary macrophage activation after release of necrotic marrow contents into the circulation.[6,7,9,10]
Diagnostic Crossroad: Sepsis and Thrombotic Microangiopathy. The initial suspicion of infection was understandable given the presence of fever and pulmonary infiltrates. Fever and pulmonary infiltrates, by contrast, are not specific, since they may be present in sepsis, ACS, and FES alike.[6] BMN/FES is frequently misinterpreted initially as infection or multiorgan failure syndrome.[8] For this reason, dynamic interpretation of laboratory trends is often more informative than isolated values.
However, the early biochemical profile (disproportionate LDH and CK elevation preceding overt inflammatory escalation) was not entirely consistent with isolated sepsis. Thrombotic microangiopathy was a legitimate alternative diagnosis. BMN/FES can closely mimic TTP, presenting with anaemia, thrombocytopenia, neurological impairment, renal dysfunction, and elevated LDH.[10] Nevertheless, preserved ADAMTS13 activity and the absence of schistocytes made classical TTP unlikely.
Extensive BMN results in the release of fat globules and necrotic marrow elements into the circulation, leading to pulmonary and systemic microvascular obstruction.[5,8] These embolic phenomena account for the characteristic combination of respiratory failure, encephalopathy, and multiorgan dysfunction. A brain MRI demonstrating the starfield pattern is a radiological hallmark of cerebral fat embolism and has been consistently described in SCD-associated FES.[6,11]
Brain MRI played a pivotal role in diagnosis. The characteristic "starfield" pattern is a major radiological clue to cerebral fat embolism and may be present even when initial brain computed tomography is normal.[7,12] Recent case reports have also highlighted that cerebral FES may manifest with severe neurological phenotypes, including status epilepticus, reinforcing the need for early MRI in unexplained neurological deterioration in sickle syndromes.[12]
Historical mortality of BMN/FES approached 64% overall.[8] However, outcomes differ significantly depending on therapeutic strategy, reaching 91% without transfusion.[8] Exchange transfusion likely improves haemodynamic rheology and limits ongoing sickling and marrow injury;[5,8] when performed early, it has been associated with improved survival by limiting bone marrow injury.[6,7,9] PEX has been used as adjunctive therapy in severe cases, with the rationale of attenuating the inflammatory cascade and removing circulating toxic mediators; however, the evidence remains limited to case reports and small series.[8,10,12] In our patient, sequential use of red cell exchange and PEX was followed by progressive haematological and neurological recovery, in line with the emerging experience reported in recent literature.[7,11,13]
Overall, this case underlines that BMN/FES should be suspected whenever a patient with sickle cell disease develops sudden multiorgan deterioration after an apparently standard VOC, especially in the presence of erythroblastosis, thrombocytopenia, marked LDH elevation, and unexplained neurological impairment. Early referral to a specialised haemoglobinopathy centre can be crucial for both diagnostic accuracy and timely initiation of exchange-based therapy.
Key Diagnostic Lessons from This Case
In sickle cell disease, sudden respiratory and neurological worsening after an apparently typical VOC crisis should prompt immediate diagnostic reassessment rather than being attributed to infection alone. In this setting, marked erythroblastosis, a rapid decline in platelet count, disproportionate LDH elevation, and hyperferritinaemia are important clues suggesting bone marrow necrosis with fat embolism syndrome. The absence of schistocytes together with ADAMTS13 activity above 10% helps distinguish this presentation from classical thrombotic thrombocytopenic purpura. Brain MRN may provide decisive support, particularly when the characteristic “starfield” pattern is present, indicating cerebral fat embolism. Early recognition is crucial because management depends on rapid specialist involvement and prompt exchange transfusion, while adjunctive plasma exchange may be considered in severe cases or when the clinical course remains refractory.References
- Zaidi AU, Glaros AK, Lee S, Wang T, Bhojwani
R, Morris E, Donohue B, Paulose J, Iorga ŞR, Nellesen D. A systematic
literature review of frequency of vaso-occlusive crises in sickle cell
disease.Orphanet J Rare Dis.2021;16(1):460. https://doi.org/10.1186/s13023-021-02096-6 PMid:34727959 PMCid:PMC8561926
- Koehl JL, Koyfman A, Hayes BD, Long B. High risk
and low prevalence diseases: Acute chest syndrome in sickle cell
disease. Am J Emerg Med.2022;58:235-244. https://doi.org/10.1016/j.ajem.2022.06.018 PMid:35717760
- Tsitsikas DA, Mihalca D, Hall J, May JE, Gangaraju
R, Marques MB, Scully M. Pitfalls in diagnosing thrombotic
thrombocytopenic purpura in sickle cell disease. J Clin
Med.2022;11(22):6676. https://doi.org/10.3390/jcm11226676 PMid:36431152 PMCid:PMC9696110
- Vichinsky EP. Comprehensive care in sickle cell
disease: its impact on morbidity and mortality. Semin
Hematol.1991;28(3):220-226.
- Ataga KI, Orringer EP.Bone marrow necrosis in
sickle cell disease: A description of three cases and a review of the
literature. Am J Med Sci. 2000. https://doi.org/10.1097/00000441-200011000-00009 PMid:11093689
- Alkindi S, Al-Musalhi M, Pathare A, et al. Fat
embolism syndrome in sickle cell disease: A single-centre experience
and review of outcomes. Sci Rep. 2025;15:11983.
- Salam A, Sameer R, Al-Ajmi E, Al-Farsi K, Khan H,
Unnisa NK, et al. Clinical, laboratory, radiological features, and
outcome of acute fat embolism syndrome in sickle cell disease. Sci Rep.
2025;15(1):27523. https://doi.org/10.1038/s41598-025-11983-y PMid:40721454 PMCid:PMC12304287
- Tsitsikas DA, Gallinella G, Patel S, Seligman H,
Greaves P, Amos RJ. Bone marrow necrosis and fat embolism syndrome in
sickle cell disease: increased susceptibility of patients with non-SS
genotypes and a possible association with human parvovirus B19
infection. Blood Rev. 2014 Jan;28(1):23-30. Epub 2014 Jan 11. https://doi.org/10.1016/j.blre.2013.12.002 PMid:24468004
- Tsitsikas DA, Vize J, Abukar J. Fat Embolism Syndrome in Sickle Cell Disease. J Clin Med. 2020 Nov 8;9(11):3601. https://doi.org/10.3390/jcm9113601 PMid:33171683 PMCid:PMC7695297
- Gangaraju R, Reddy VV, Patel K, et al. Fat
embolism syndrome due to bone marrow necrosis in patients with sickle
cell disease: Diagnostic challenges and therapeutic considerations. Am
J Hematol. 2019;94(2):E46-E49. https://doi.org/10.1002/ajh.25363 PMid:30479040
- Rizvi S, Khakwani M, Pancham S, Tsitsikas D,
Rudzki Z, Hassan-Smith G, Bowen M, Wright C, Park D. Bone marrow
necrosis and fat embolism syndrome in sickle cell disease during
COVID-19 infection treated successfully with sequential red cell and
plasma exchange. EJHaem. 2022 Dec 15;4(1):207-10. https://doi.org/10.1002/jha2.621 PMid:36718354 PMCid:PMC9877825
- Bortolotti M, Costamagna G, Gagliardi D, Migone De
Amicis M, Bresolin N, Graziadei G. A suspected case of cerebral fat
embolism triggering a drug-resistant status epilepticus in a
HbS/beta+-thalassaemia patient. Mediterr J Hematol Infect Dis.
2022;14(1):e2022019. https://doi.org/10.4084/MJHID.2022.019 PMid:35444771 PMCid:PMC8992611
- Alsaghir A, Alsaghir L, Alsaif J, Mobeireek A.
Successful therapeutic plasma exchange for a patient with sickle cell
disease and fat embolism syndrome after a failure of a response to red
cell exchange transfusion. Transfusion. 2023;63 Suppl 1:S33-S36. https://doi.org/10.1111/trf.17220 PMid:36748667