A 20-year-old male, born of a consanguineous marriage with no known comorbidities, presented with a month of high-grade, recurrent fever, chills, loose stools, abdominal pain, loss of appetite, and unintentional weight loss. Physical examination revealed marked splenomegaly (24cm) and generalised lymphadenopathy (the largest being 2cm). Initial laboratory work-up showed pancytopenia (hemoglobin 10.8 g/dL, total leukocyte count 4,900/µL with severe neutropenia at 10%, and platelet count 27,000/µL) and elevated liver enzymes. Extensive testing for infectious causes was negative. Further investigations showed markedly elevated serum ferritin (4868.50 ng/ml) and Lactate Dehydrogenase (1343 U/l, normal range 120-246 U/l). Bone marrow aspiration showed an increased number of lymphocytes (40%) and hemophagocytosis by macrophages. (fulfilling 5 out of 8 required criteria according to HLH 2004, as shown in Table 1).[5] A contrast-enhanced CT scan of the neck, chest and abdomen revealed hepatosplenomegaly and widespread non-necrotic lymphadenopathy, prompting a cervical lymph node biopsy. Histopathology showed effacement of nodal architecture by scattered large atypical mononuclear cells in a mixed inflammatory background. Immunohistochemistry (CD30+, CD15+ (subset), weak PAX5+, MUM1+, CD20-, CD3-, CD45-, OCT2-, EMA-) confirmed classic Hodgkin lymphoma (CHL) (Figure 1). Simultaneously, bone marrow biopsy revealed hypercellular marrow (80–90%) involved by CHL, along with a marked increase in histiocytes exhibiting prominent hemophagocytosis and emperipolesis, consistent with HLH (Figure 2). The patient was started on dexamethasone and with 50% dose of etoposide (as per HLH-2004 protocol) along with ABVD chemotherapy. After cycle one, the fever resolved, blood counts normalised, and splenic size decreased, and he was continued on lymphoma-directed therapy. Given the advanced disease, brentuximab vedotin (BV) was added; the remaining 5 BV-AVD cycles were completed uneventfully. The combination of gross splenomegaly and HLH prompted testing for an underlying genetic predisposition. Whole-exome sequencing identified a pathogenic STXBP2 splice-site mutation (c.1247-1G>C), diagnostic of FHL5, autosomal recessive inheritance. The patient and the family were counselled regarding the necessity of an allogeneic bone marrow transplant, but they decided to postpone the treatment, citing socioeconomic reasons.
 |
Table 1. Diagnostic criteria of hemophagocytic lymphohistiocytosis: HLH-2004 4. |
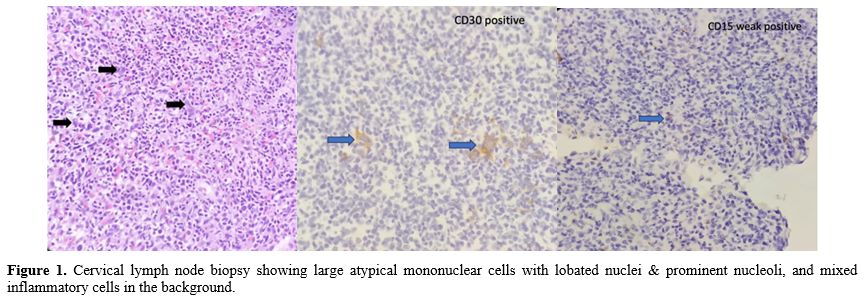 |
Figure 1.
Cervical lymph node biopsy showing large atypical mononuclear cells
with lobated nuclei & prominent nucleoli, and mixed inflammatory
cells in the background. |
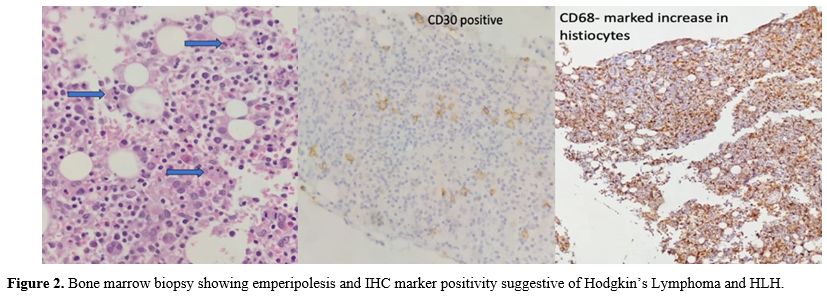 |
Figure 2. Bone marrow biopsy showing emperipolesis and IHC marker positivity suggestive of Hodgkin’s Lymphoma and HLH. |
Six months after the primary therapy, the patient experienced a recurrence of fever, cytopenia, and systemic symptoms. His blood work suggested a relapse of HLH. Bone marrow and PET-CT showed no evidence of lymphoma recurrence. The patient responded to the re-initiated HLH-2004 protocol. HLA typing and clinical exome sequencing were performed for family members. A matched unrelated donor search failed. A 10/10 HLA-matched brother was homozygous for the STXBP2 mutation, so a 7/10 HLA-matched, heterozygous sibling was chosen as the donor. The conditioning regimen was Fludarabine-Melphalan-Antithymocyte Globulin, with Post-Transplant Cyclophosphamide + Calcineurin Inhibitors + Mycophenolate Mofetil for GVHD prophylaxis. Infection risks were managed with a prophylactic regimen comprising intravenous immunoglobulin and dual antifungal therapy. Post-transplant complications included SARS-CoV-2 and Enteropathogenic E. coli infections. Leucocyte and platelet engraftment occurred on days +28 and +27, respectively. Chimerism was 100% donor at day +237.
The STXBP2 (Syntaxin Binding Protein 2) gene mutation causes FHL5, an autosomal recessive disorder that impairs the cytolytic function of NK cells and CD8+ T lymphocytes, which is crucial for controlling viral infections and limiting immune system overactivity. These defects lead to uncontrolled immune activation and subsequent hypercytokinemia.[6]
The association of STXBP2 mutation with malignancy, along with HLH, is limited to a few case reports. A Norwegian case report by Machaczka et al described a case of a 17-year-old girl with a biallelic STXBP2 mutation who developed life-threatening HLH and was treated with HLH-94 protocol with subsequent monthly IVIg due to hypogammaglobulinemia. Forty-five months after initial diagnosis, the patient presented with rapidly progressive unilateral neck swelling, which was diagnosed as Hodgkin’s Lymphoma. She was treated with ABVD, IFRT and HSCT.[7] We came across another case from Pakistan by Mirza et al, with a presentation similar to our case. A 23-year-old male patient, initially diagnosed and treated as Hodgkin’s Lymphoma, went on to develop HLH after a year. He was found to have two pathogenic variants in the STXBP2 gene and was treated with allo-SCT with a full matched sibling, harbouring the same heterozygous mutation as the donor.[8] Heterozygous STXBP2 mutations have been identified in pediatric patients with Langerhans cell Histiocytosis[9] and acute leukaemia[10] who also presented with HLH.
A recently published Chinese study investigated whether germline mutations in FHL-related genes predispose individuals to mature T- and natural killer (NK)-cell lymphomas, specifically peripheral T-cell lymphoma (PTCL). Whole-exome and targeted next-generation sequencing of paired germline and tumour DNA from 74 Chinese patients with T/NK-cell lymphomas revealed FHL-related germline mutations (UNC13D, STXBP2, PRF1, and STX11) in 18.9% of patients, despite no prior history of HLH or immune dysfunction.[11]
This case contributes to the limited literature on STXBP2-associated disease within the context of HLH and malignancy. Early recognition and treatment are essential to improving survival outcomes. Genomic testing for related mutations is recommended for adolescents and young adults with malignancy-associated HLH. Identification of these mutations can alter the treatment decisions, including the potential for hematopoietic stem cell transplantation (HSCT).
References
- Joanne I. Hsu, Sarah Nikiforow, Nancy Berliner; Hemophagocytic lymphohistiocytosis in adults. Blood 2026; 147 (10): 1037–1047. https://doi.org/10.1182/blood.2025031100
- Pai SY, Lurain K, Yarchoan R. How immunodeficiency
can lead to malignancy. Hematology Am Soc Hematol Educ Program. 2021
Dec 10;2021(1):287-295. https://doi.org/10.1182/hematology.2021000261 PMID: 34889385; PMCID: PMC8791117
- Tralongo P., Bakacs A., Larocca L.M. EBV-related
lymphoproliferative diseases: a review in light of new classifications.
Mediterr J Hematol Infect Dis 2024, 16(1): e2024042, http://dx.doi.org/10.4084/MJHID.2024.042
- Haas OA (2019) Primary Immunodeficiency and Cancer
Predisposition Revisited: Embedding Two Closely Related Concepts Into
an Integrative Conceptual Framework. Front. Immunol. 9:3136. https://doi.org/10.3389/fimmu.2018.03136
- Kim, Yu & Kim, Dae-Young. (2021). Current
status of the diagnosis and treatment of hemophagocytic
lymphohistiocytosis in adults. BLOOD RESEARCH. 56. S17-S25. https://doi.org/10.5045/br.2021.2020323
- Zur Stadt, U., Schmidt, S., Kasper, B., Beutel, K., Diler, A. S., Henter, J. I., ... & Janka, G. (2009). Linkage of familial hemophagocytic lymphohistiocytosis (FHL) type 5 to chromosome 19p and identification of mutations in the STXBP2 gene. Human Molecular Genetics, 18(2), 631-639.
- Machaczka M, Klimkowska M, Chiang SC, Meeths M,
Müller ML, Gustafsson B, Henter JI, Bryceson YT. Development of
classical Hodgkin's lymphoma in an adult with biallelic STXBP2
mutations. Haematologica. 2013 May;98(5):760-4. https://doi.org/10.3324/haematol.2012.07309 PMID: 23100279; PMCID: PMC3640121.
- Mirza Rameez Samar, Daania Shoaib, Nida e Zehra,
Munira Moosajee. Late-onset Familial Hemophagocytic Lymphohistiocytosis
in a survivor of Hodgkin's Lymphoma, Leukaemia Research Reports, Volume
21, 2024, 100394, ISSN 2213-0489, https://doi.org/10.1016/j.lrr.2023.100394
- Viñas-Giménez L, Rincón R, Colobran R, de la Cruz
X, Celis VP, Dapena JL, Alsina L, Sayós J, Martínez-Gallo M (2021).
Case Report: Characterising the Role of the STXBP2-R190C Monoallelic
Mutation Found in a Patient With Hemophagocytic Syndrome and Langerhans
Cell Histiocytosis. Front. Immunol. 12:723836. https://doi.org/10.3389/fimmu.2021.723836
- Liao, Meiling MM; Yu, Jie MD. Secondary Leukaemia
in a Patient With EBV-HLH Carrying a Heterozygous STXBP2 Variant.
Journal of Pediatric Haematology/Oncology 44(2): p. e526-e528, March
2022. https://doi.org/10.1097/mph.0000000000002141
- Wei C, Zhang Y, Zhao D, Zhang W, Zhou D. Germline
defects of familial haemophagocytic lymphohistiocytosis—Related genes
may represent a predisposing factor for mature T- and natural
killer-cell lymphoma. Br J Haematol. 2025; 207(3): 842–850. https://doi.org/10.1111/bjh.20231