
HEMATOPOIETIC STEM CELL TRNSPLANTATION IN THALASSEMIA AND RELATED DISORDERS
Main Article Content
Keywords
Anemia, Thalassemic syndromes, Hemopoietic Stem Cell Transplantation
Abstract
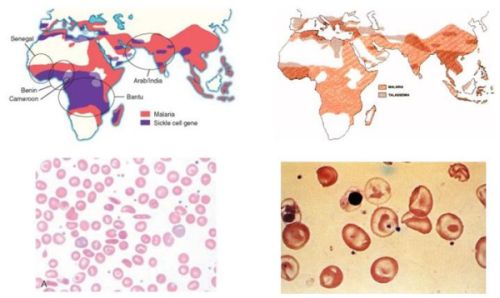
The basis of allogeneic hemopoietic stem cell (HSC) transplantation in thalassemia consists in substituting the ineffective thalassemic erythropoiesis with and allogeneic effective one. This approach is an efficient way to obtain a long lasting, probably permanent, clinical effective correction of the anaemia avoiding transfusion requirement and subsequent complications like iron overload. The first HSC transplant for thalassemia was performed in Seattle on Dec 2, 1981. In the early eighties transplantation procedure was limited to very few centres worldwide. Subsequently between 17 December 1981 and 31 January 2003, over 1000 consecutive patients, aged from 1 to 35 years, underwent transplantation in Pesaro. After the pioneering work by the Seattle and Peasaro groups, this therapeutic approach is now widely applied worldwide.
Medical therapy of thalassemia is one of the most spectacular successes of the medical practice in the last decades. In recent years advances in knowledge of iron overload patho-physiopathology, improvement and diffusion of diagnostic capability together with the development of new effective and safe oral chelators promise to further increase success of medical therapy. Nevertheless situation is dramatically different in non-industrialized countries were the very large majority of patients live today . Transplantation technologies have improved substantially during the last years and transplantation outcome is likely to be much better today than in the ‘80s. Recent data indicated a probability of overall survival and thalassemia free survival of 97% and 89% for patients with no advanced disease and of 87% and 80% for patients with advanced disease. Thus the central role of HSC in thalassemia has now been fully established. Thalassemia remains the only definitive curative therapy for thalassemia and other hemoblobinopathies. The development of oral chelators has not changed this position. However this has not settled the controversy on how this curative but potentially lethal treatment stands in front of medical therapy for adults and advanced disease patients. In sickle cell disease HSC transplantation currently is reserved almost exclusively for patients with clinical features that indicate a poor outcome or significant sickle-related morbidity.
Downloads
Abstract 913
PDF Downloads 405
HTML Downloads 3879
Article Sidebar
Article Details
How to Cite
This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc/4.0) which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Transfer of Copyright and Permission to Reproduce Parts of Published Papers. Authors will grant copyright of their articles to the Institute of Hematology, Catholic University, Rome . No formal permission will be required to reproduce parts (tables or illustrations) of published papers, provided the source is quoted appropriately and reproduction has no commercial intent. Reproductions with commercial intent will require written permission and payment of royalties.